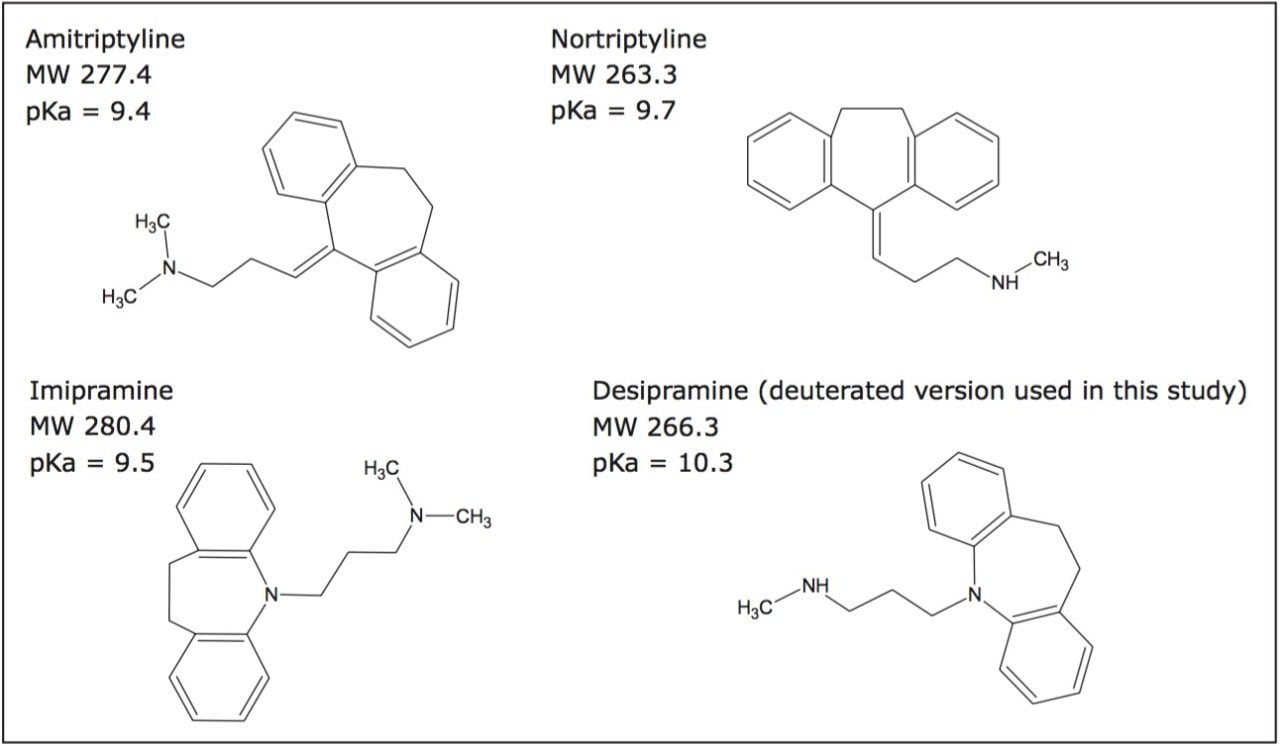
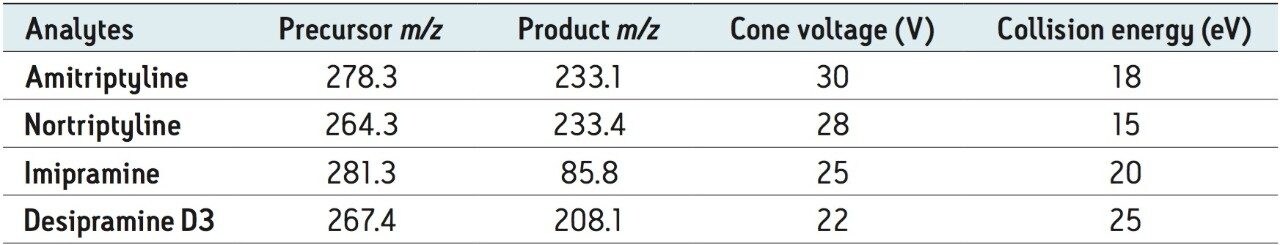
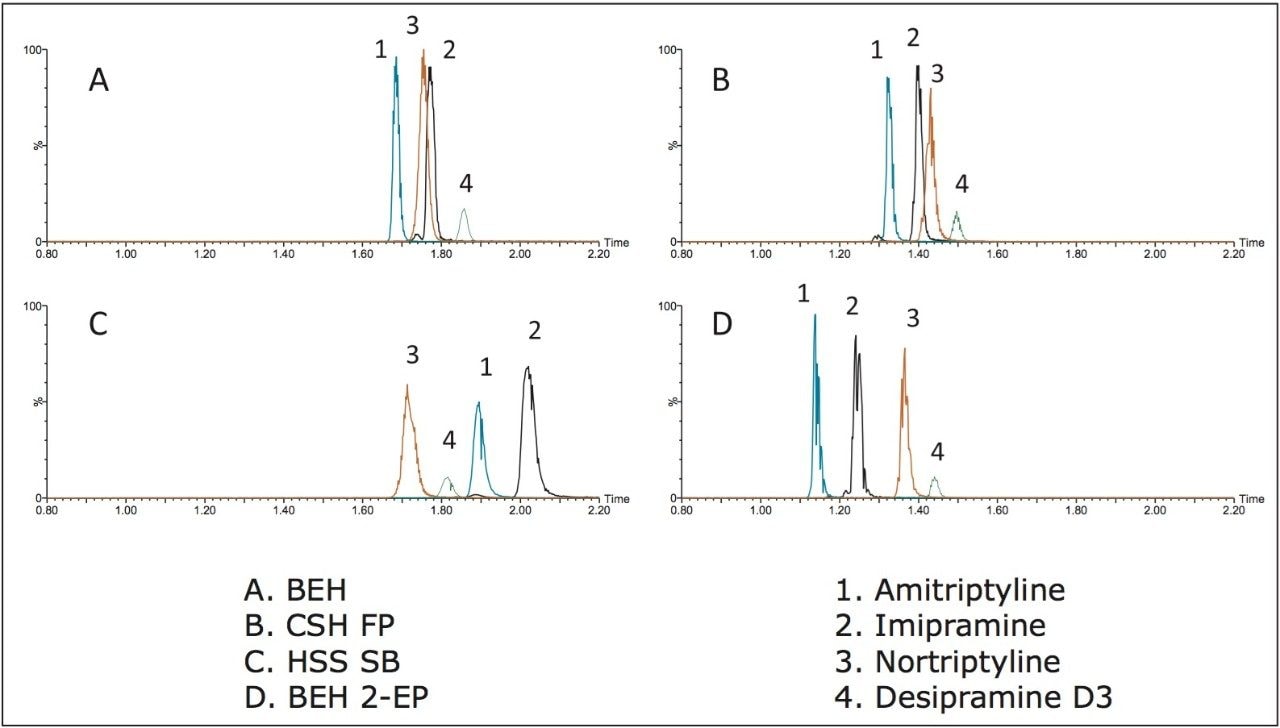
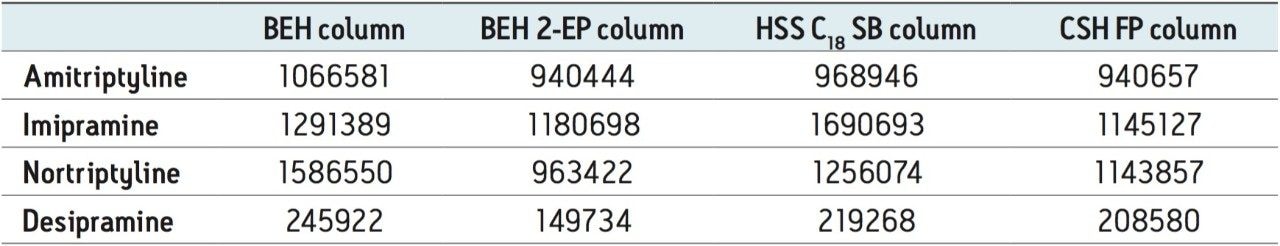
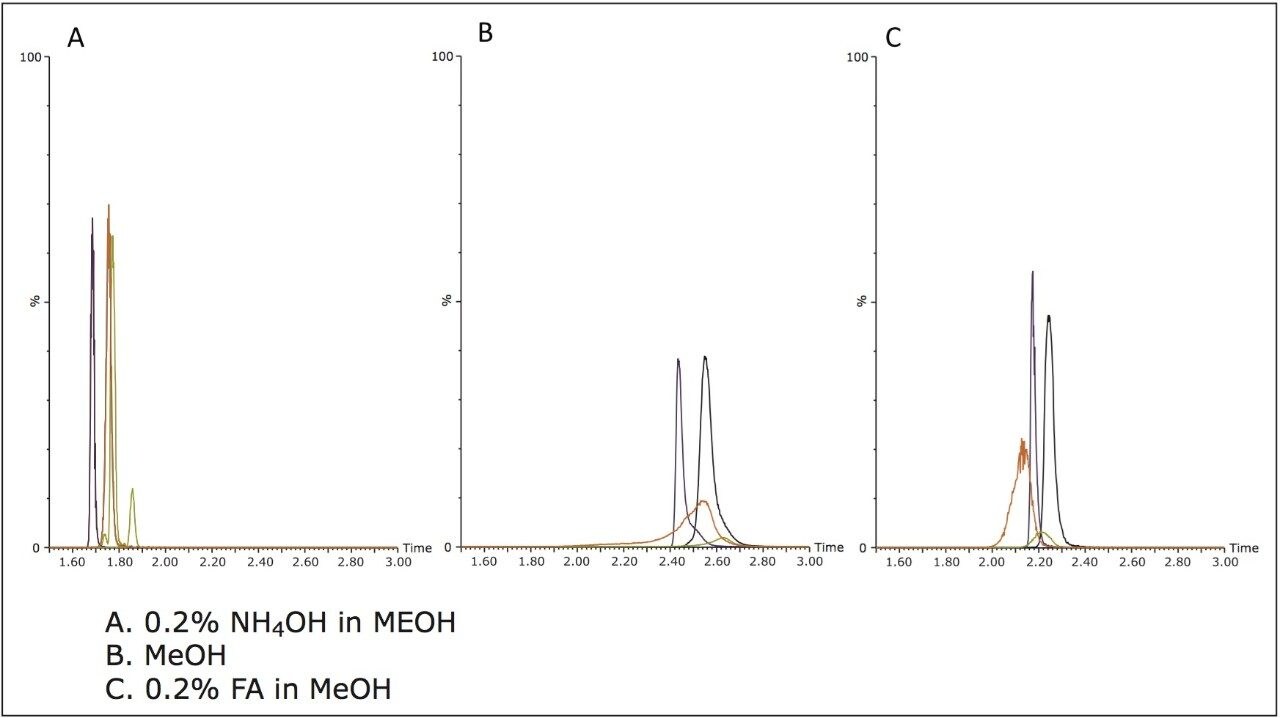
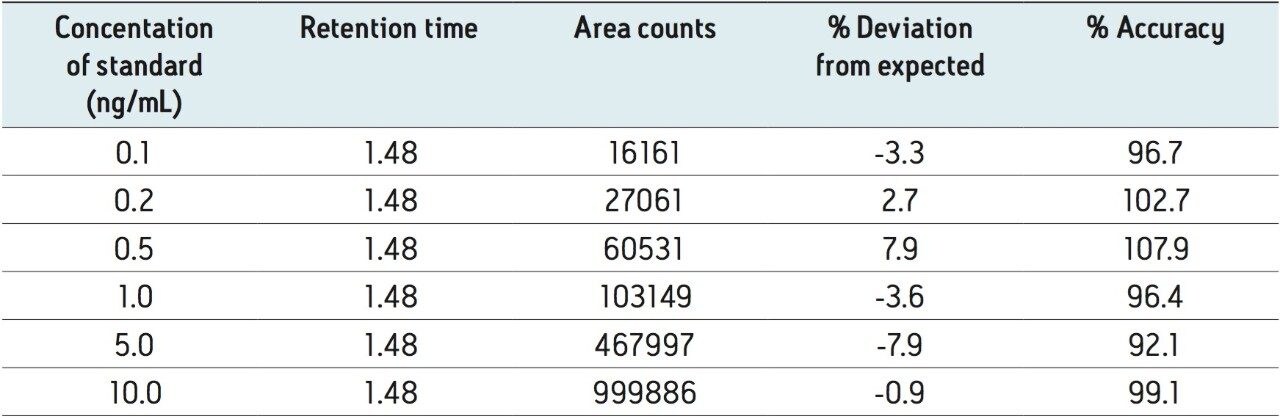
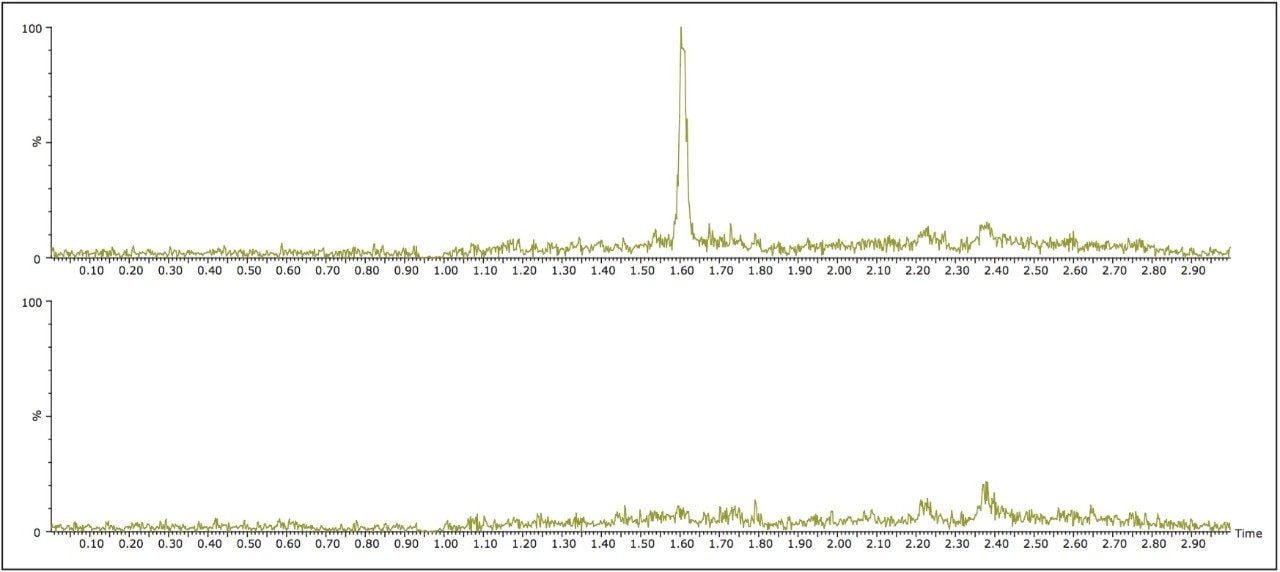
Although tricyclic antidepressants (TCAs) are an older class of drug, they remain pharmacologically relevant. For example, inadequate response to other classes of antidepressants in dosed subjects remains common, ensuring the continued viability of TCAs. Furthermore, the fact that they are significantly less expensive than some of the newer antidepressants warrants their use in outpatient clinics. Historically, TCAs have been analyzed using GC,1 UV,2 or LC-MS.3 UPC2 technology is particularly attractive for bioanalytical assays due to its compatibility with the organic extracts produced by the most common sample preparation techniques utilized in this area, such as protein precipitation (PPT), liquid-liquid extraction (LLE), and solid phase extraction (SPE). The non-aqueous solvent system does not require evaporation and reconstitution of organic extracts prior to injection in the manner often necessary in reversed phase chromatography. Characterized by the use of an environmentally sustainable, renewable primary solvent (CO2), orthogonality to reversed phase chromatography, and availability of diverse stationary phases, UPC2 provides the benefits of reduced solvent consumption and cost, increased bioanalytical method selectivity, and flexibility in method development. Therefore, a proof of concept study was performed to determine the applicability of UPC2 separations to the analysis of TCAs in human urine, as a representative methodology. Structures for the specific TCAs used in the study are shown in Figure 1. While method development approaches for GC and LC are well defined and routinely used, method development for UPC2 separations is a newer area of research. This application highlights certain key UPC2 parameters and suggests appropriate screening options. For example, column chemistry, pH, and injection solvent were systematically and automatically screened using a column manager and a four-solvent switching system. Once a column, mobile phase, and injection solvent were chosen, the gradient was optimized, and extracted urine samples were analyzed using the best conditions from the screening experiments. Lower limits of quantification (LLOQs) of 0.1 ng/mL in urine were easily achieved for each of the antidepressants tested, and met the FDA criteria for LLOQ determination for bioanalytical methods. In addition, abbreviated standard curves (without internal standard) were linear and accurate within +/- 1% to 8%.