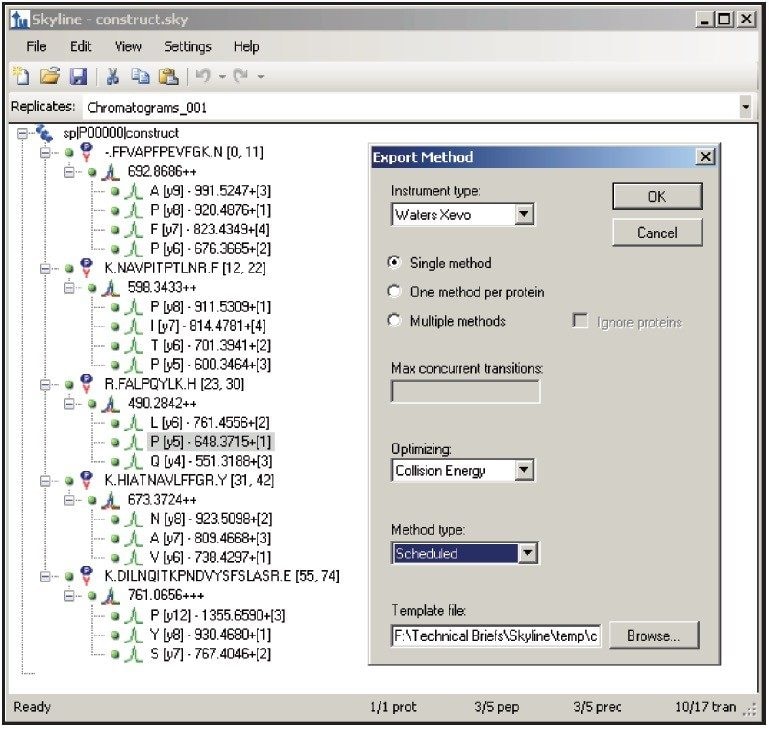
Five allergenic peptide standards were spiked in a matrix and the best candidate transitions predicted using Skyline, a software application for building SRM/MRM quantitative methods and analyzing the resulting mass spectrometer data. Skyline is vendor neutral and it was developed for creating and iteratively refining targeted methods for large-scale studies. A scheduled SRM/MRM method was created in order to independently optimize the collision energy for all peptides. The retention times were predicted with the built-in model of Skyline using the observed retention time as input/landmarks for linear retention time prediction and curve optimization/adjustment.
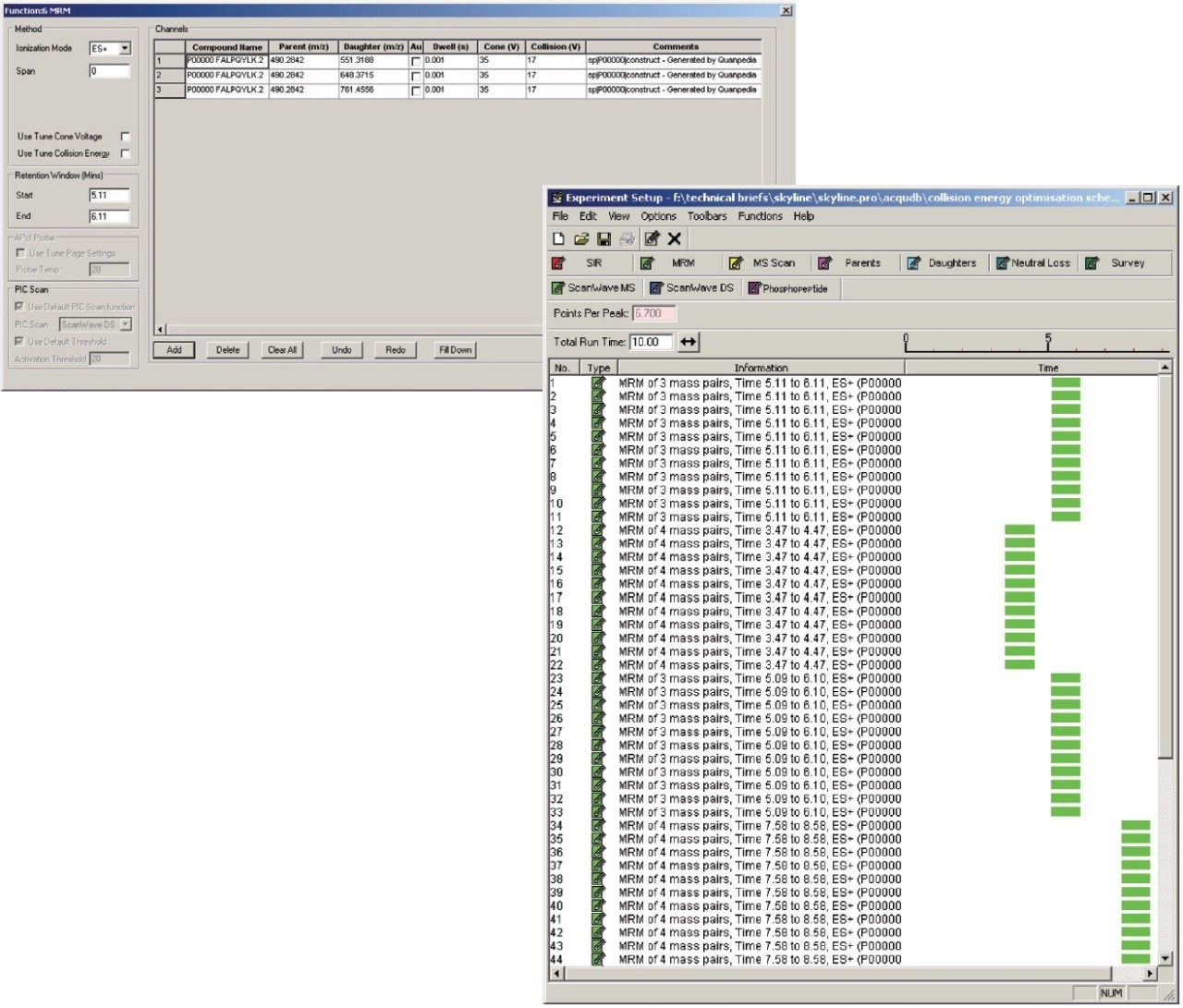
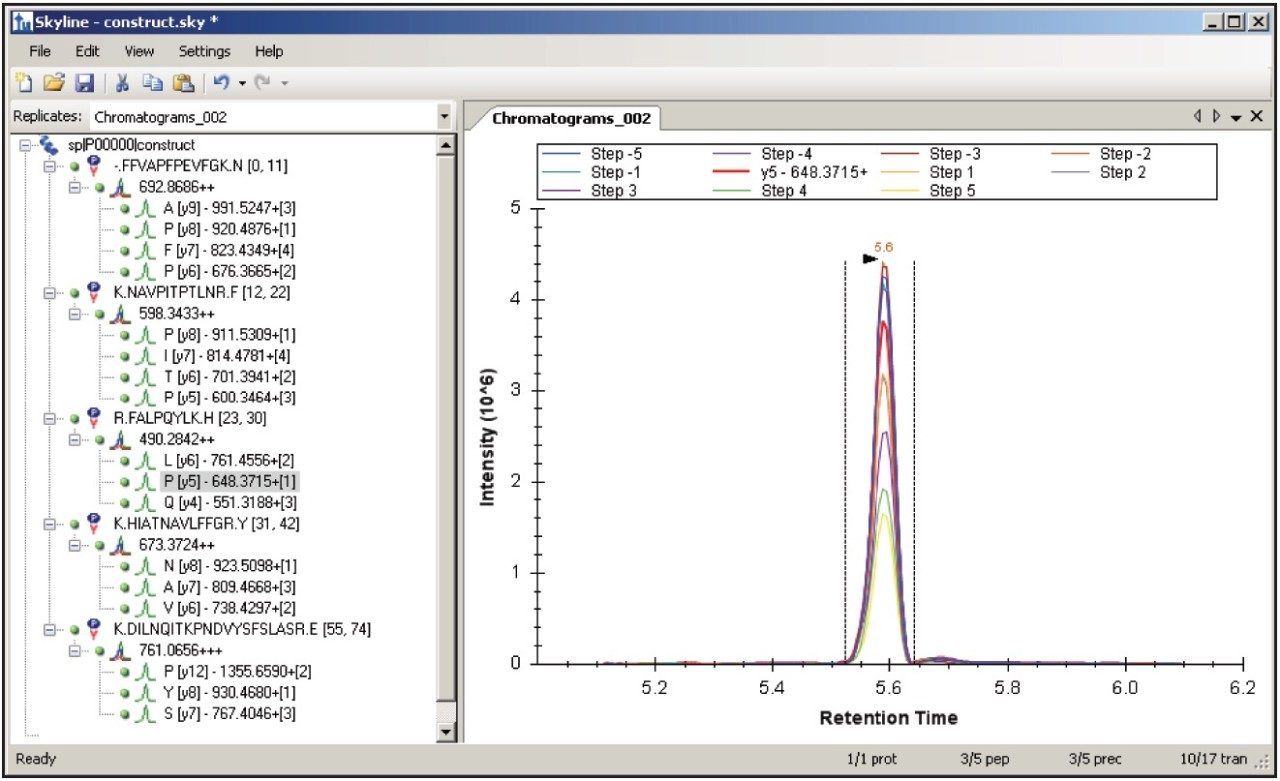
Figure 1 shows the predicted transitions for the allergenic protein of interest and the use of the Skyline Export Method editor for creating a retention time-scheduled Xevo TQ-S collision energy optimization experiment. A set of 11 collision energies was created, ranging from -5 V to +5 V, versus the optimally predicted collision energy derived from empirical observations for each peptide. The created method can be directly read and edited by the Experiment Setup SRM/MRM method editor of MassLynx Software, shown in Figure 2. The dwell times are automatically calculated based on the average chromatographic peak width. After the experiment is run, the results can either be reviewed in MassLynx or Skyline software. Figure 3 overlays the results of 11 SRM/MRM experiment for one of the peptides of interest. In this instance, the optimal collision energy was found to be 2 V lower than the predicted value and in agreement with the MassLynx Software data observed values, which were obtained via infusion of the peptide standards.