Disulfide bond formation is critical for establishing and maintaining proper three-dimensional folding and therefore functions of therapeutic proteins, e.g., monoclonal antibodies (mAbs). Localization and assignment of disulfide bonds are an important aspect of protein structural analysis.
However, the identification of disulfide pairing in a protein with multiple cysteine residues is generally more time-consuming and challenging than the determination of the protein sequence due to incomplete disulfide bond formation and disulfide bond scrambling.1,2
The combination of enzymatic digestion with liquid chromatography-mass spectrometry (LC-MS) is frequently applied as a routine method for the assignment of disulfide bonds.3,4 However, peptide mapping via an LC-MS approach is traditionally labor-intensive and time-consuming in data processing and interpretation.
Furthermore, the assignment of disulfide linkages sometime is inconclusive when a protein contains multiple cysteines due to the significantly increased number of possible disulfide-bonded peptide isomers.1 For example, there are totally 105 possible disulfide-bond pairing schemes for a protein containing eight cysteine residues (while an IgG1 mAb typically has 32 or more cysteines in its two light chains and two heavy chains).
This, plus the reality of potential disulfide bond scrambling, makes the disulfide bond assignment extremely difficult when the analysis is performed manually. An automated workflow is highly preferred for quick assessment of heterogeneities of therapeutic proteins caused by the disulfide linkages.
Recently, we have developed an integrated peptide mapping workflow for protein sequence confirmation and characterization using a UPLC-MSE method for data collection and a targeted bioinformatic tool, the BiopharmaLynx Application Manager for MassLynx Software, for automated data processing and annotation.5
The data independent MSE approach alternatively collects mass spectrometric data of precursors and fragments of eluting peptides from protein enzymatic (e.g., tryptic) digests in an unbiased manner.6 The integrated workflow overcomes most shortcomings of traditional peptide mapping methods7 and has been demonstrated to be fast, robust and suitable for routine analysis of recombinant proteins such as therapeutic mAbs5 and subunit vaccines.8 The approach has also been successfully applied to identify expected disulfide bonds in an IgG1 mAb.9
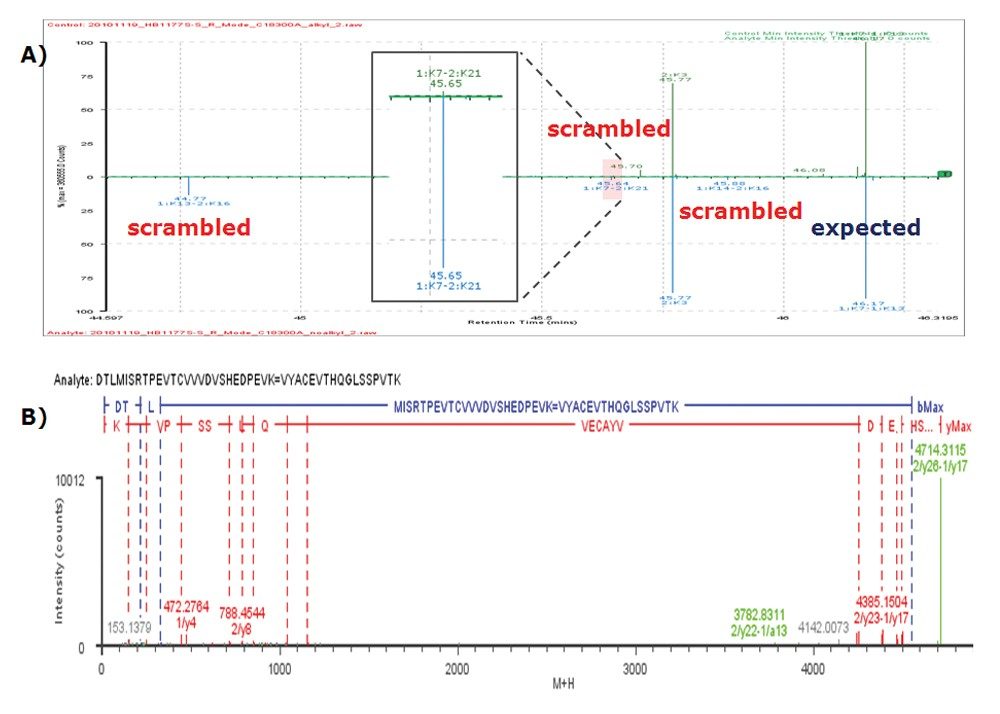
In this study, an advanced UPLC-MSE peptide mapping workflow is demonstrated for the detection and identification of disulfide linkages (including scrambled disulfide linkages) in a recombinant IgG1 mAb. The goal is to develop a viable approach for automatic mapping or monitoring of disulfide bonded peptides in a protein with multiple cysteines.
The significances of the integrated workflow include:
- Enhanced mass resolution and mass accuracy using the SYNAPT HDMS G2 QTof MS System,10 which is critical for acquiring high-quality MSE data for large disulfide-bond-linked peptides with high molecular weight.
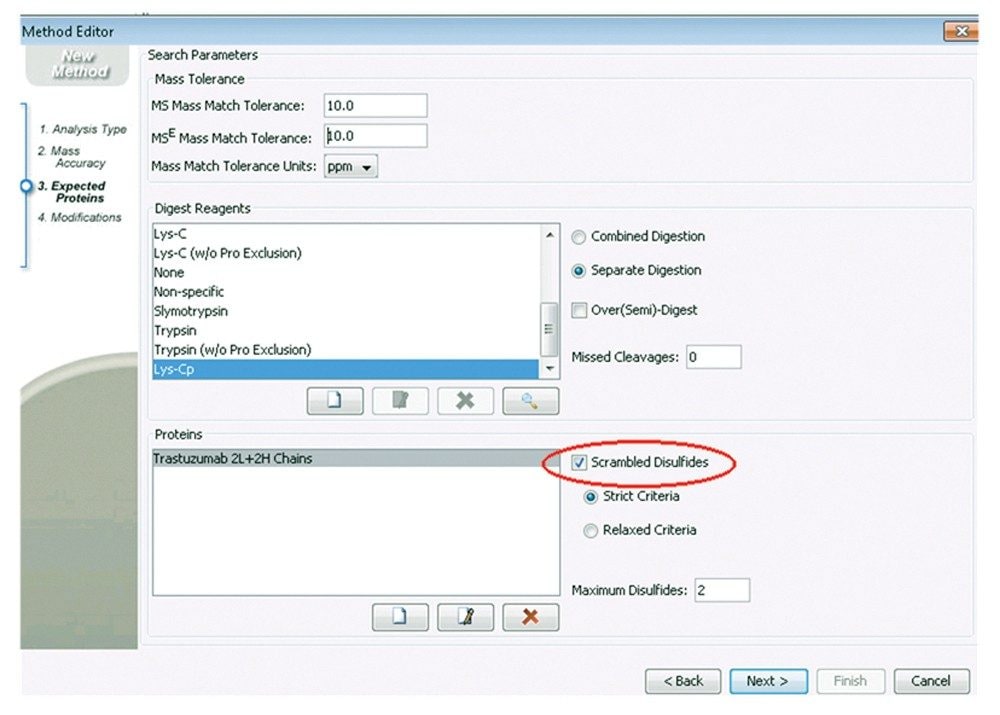
- Automatic assignment of disulfide linkages by an upgraded BiopharmaLynx (version 1.3) in a randomized mode, which is important for the identification of potential scrambled disulfide bonds in an automatic mode.
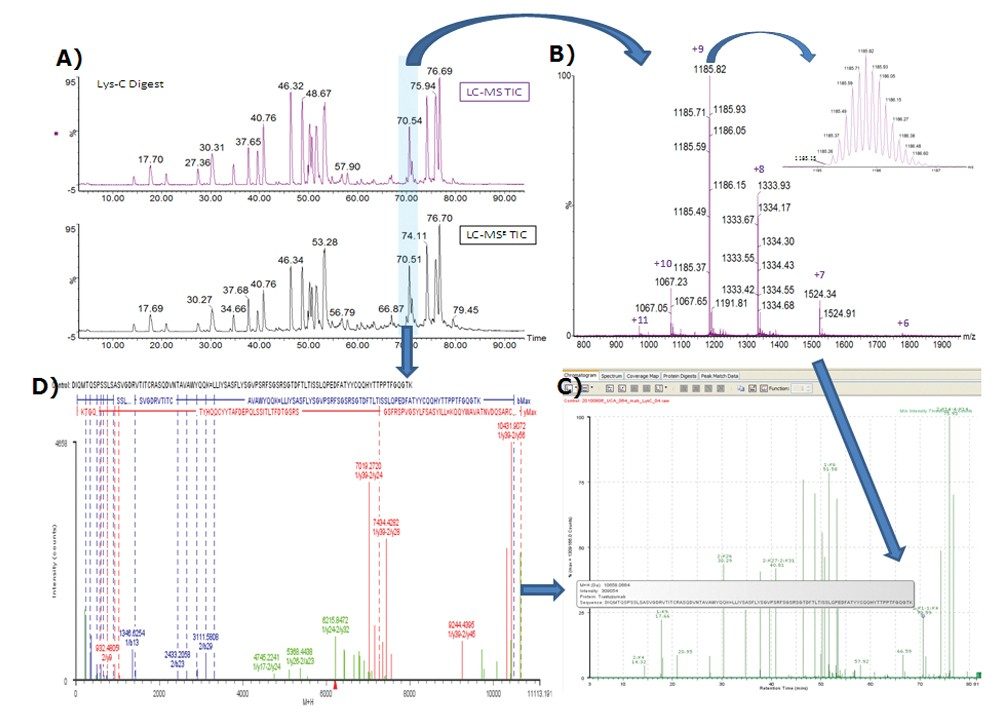
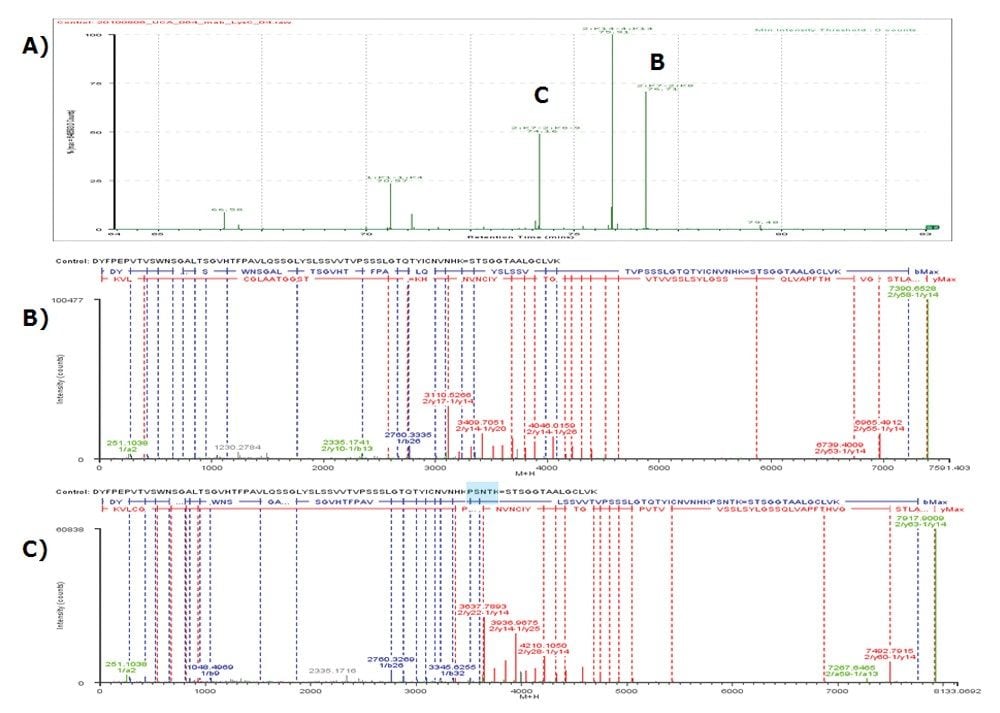
To reduce the complexity of protein digest mixture, the mAb was digested with endoproteinase Lys-C. The obtained digests were separated by reversed-phase LC with an ACQUITY UPLC System, followed by online MSE detection and BiopharmaLynx 1.3 analysis. Digests prepared with and without alkylation protection of free sulfhydryl groups2 in the sample were used to differentiate native scrambled disulfide linkages in the mAb from those potentially formed during sample preparation stage.