Generic or standard chromatographic methods in combination with orthogonal acceleration time-of-flight (oa-ToF) mass spectrometry have become critical tools for use in high-throughput QC screening of synthetic medicinal compounds. Short analysis times are often employed due to the high sample numbers required for fast lead discovery strategies. Over recent years, the combination of LC and ToF MS has proven to be a suitable analytical technique to address these needs. Furthermore, the technique provides mass accuracy within 5 ppm of the actual value which is required for journal publication, patent submission and accurate structural identification via elemental composition calculations.
The use of more conventional techniques such as NMR cannot address these high throughput analytical needs due to relatively poor sensitivity, high sample purity requirement, necessity of operator expertise and the use of costly solvents. To simplify and streamline the analytical procedures, automation in combination with open access is a key factor for this type of application.
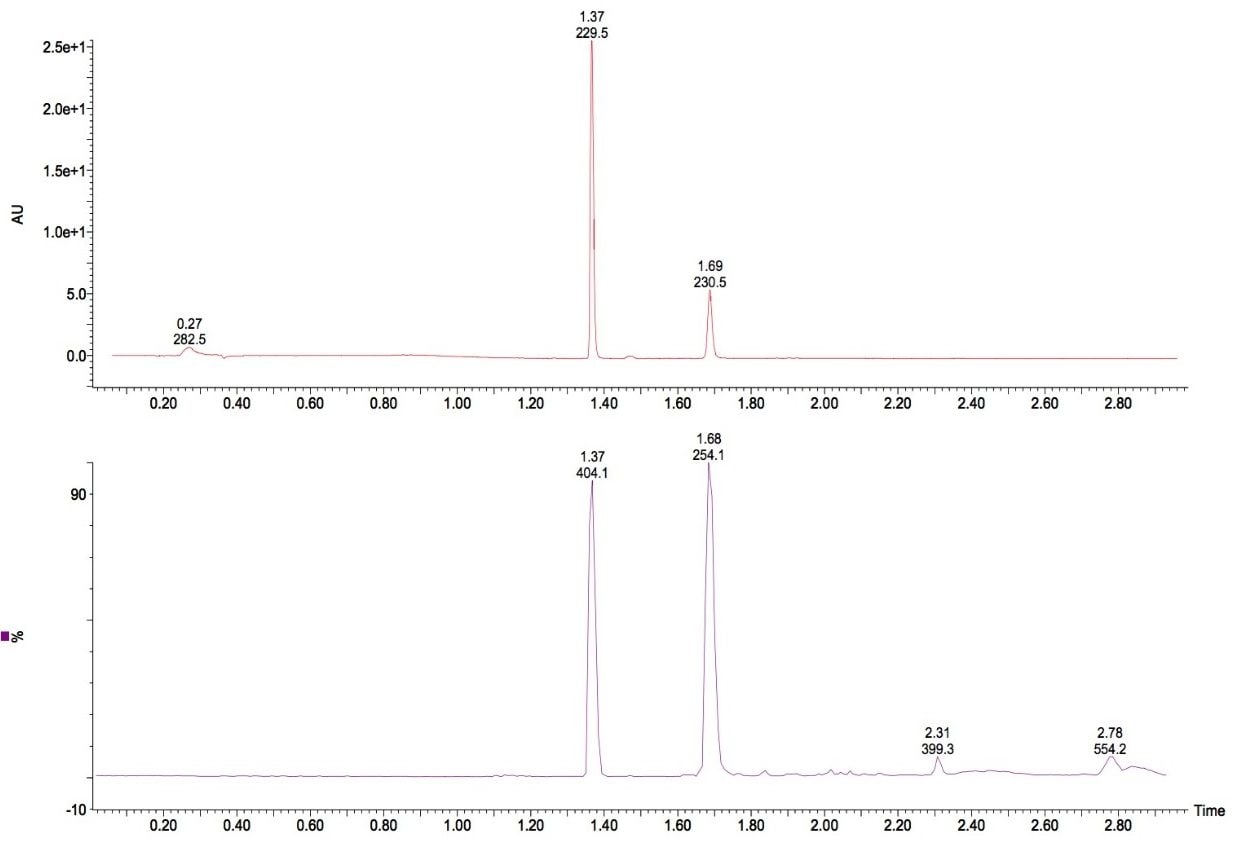
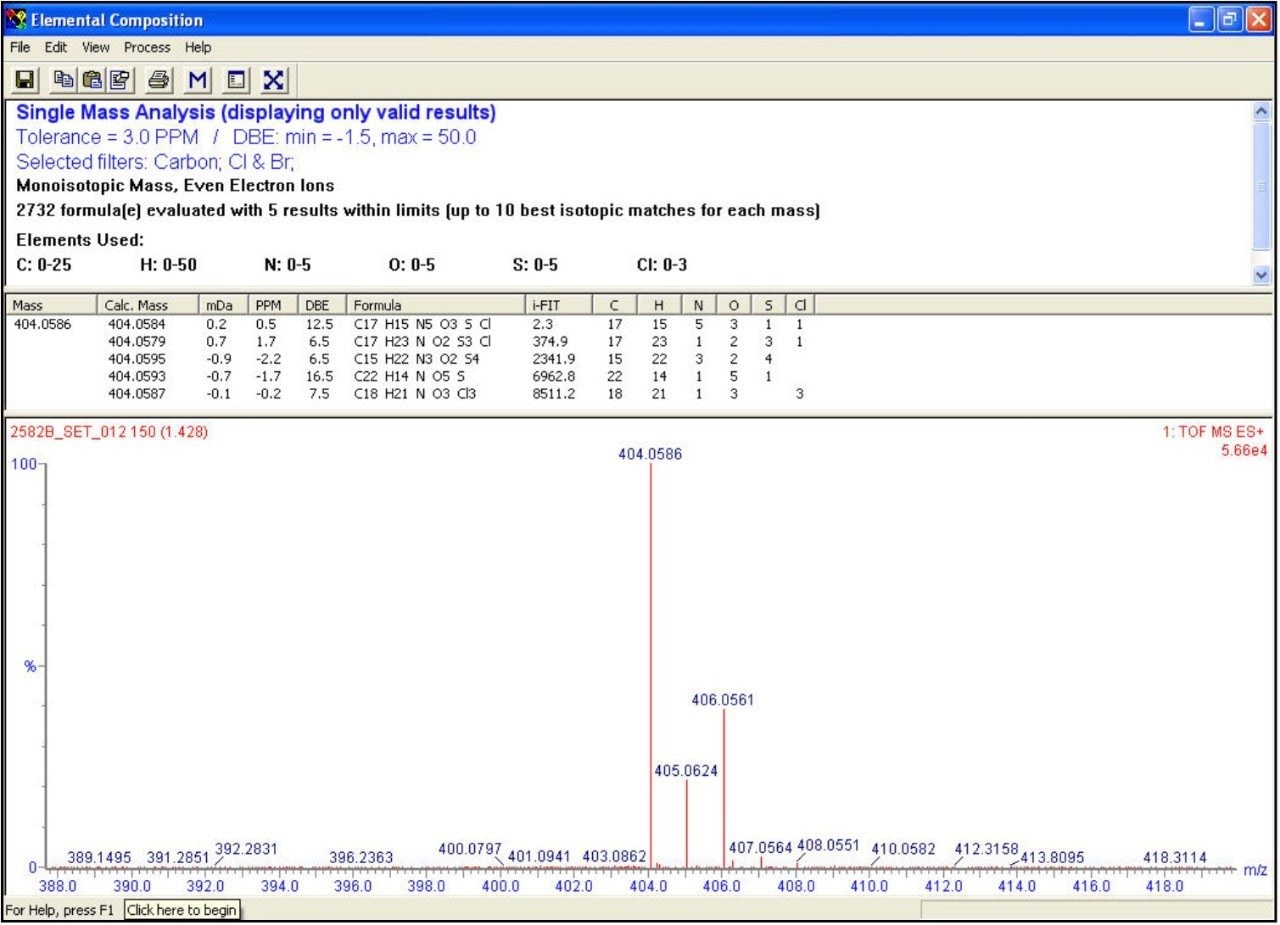
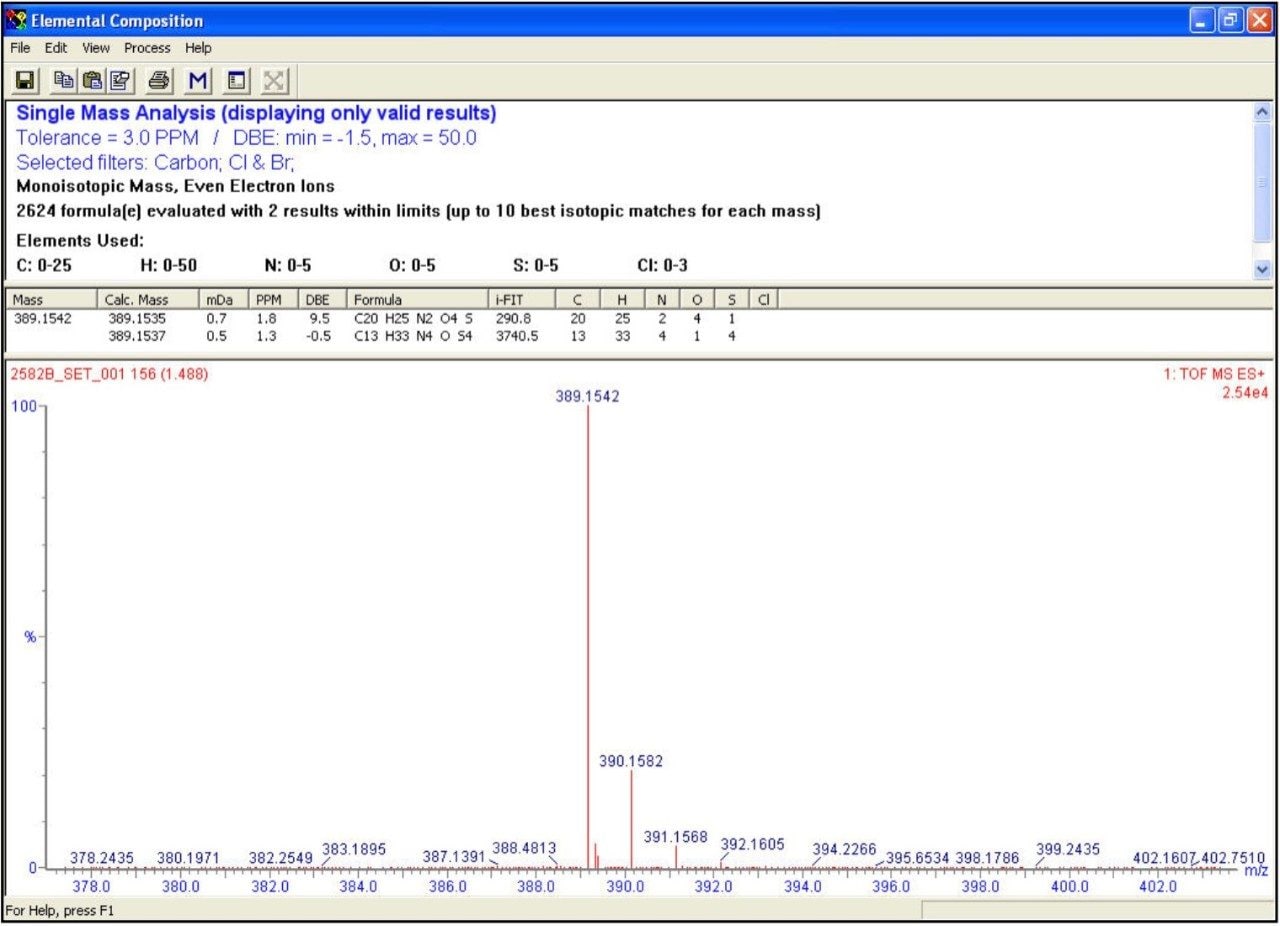
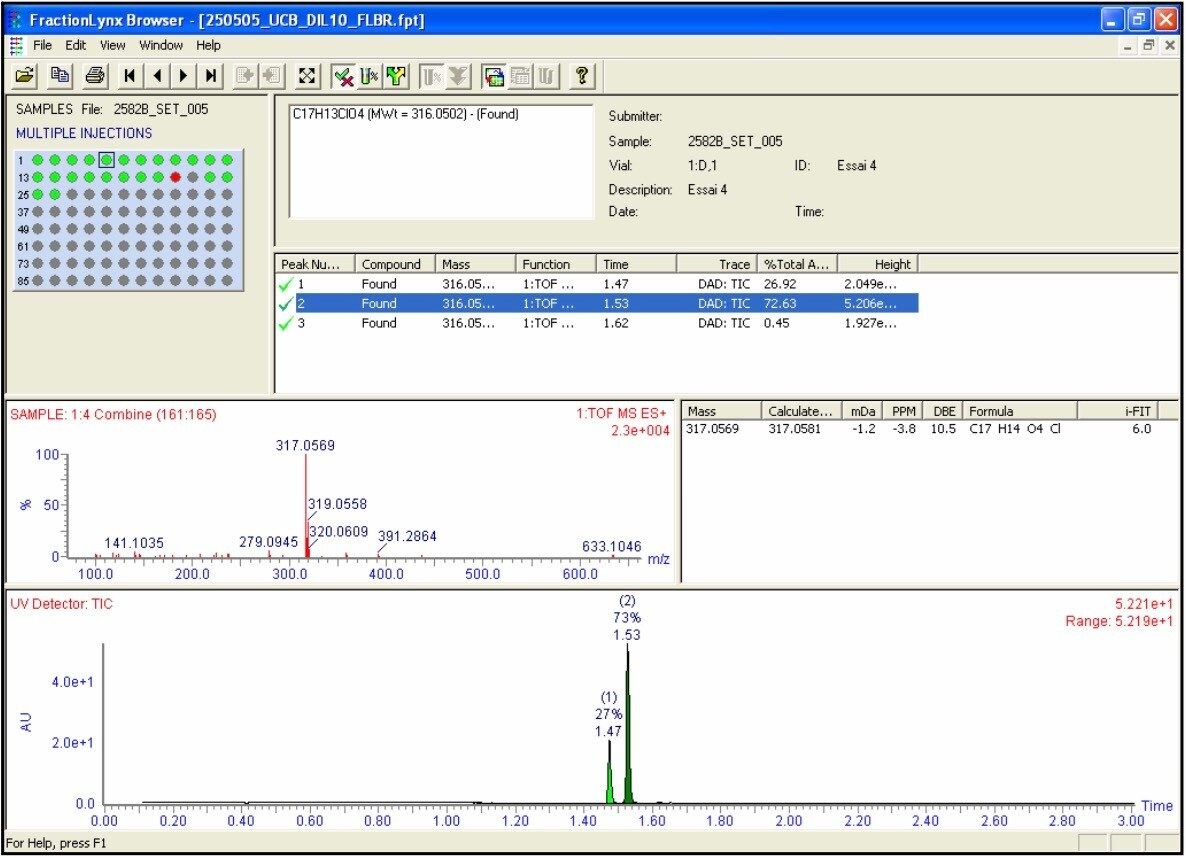
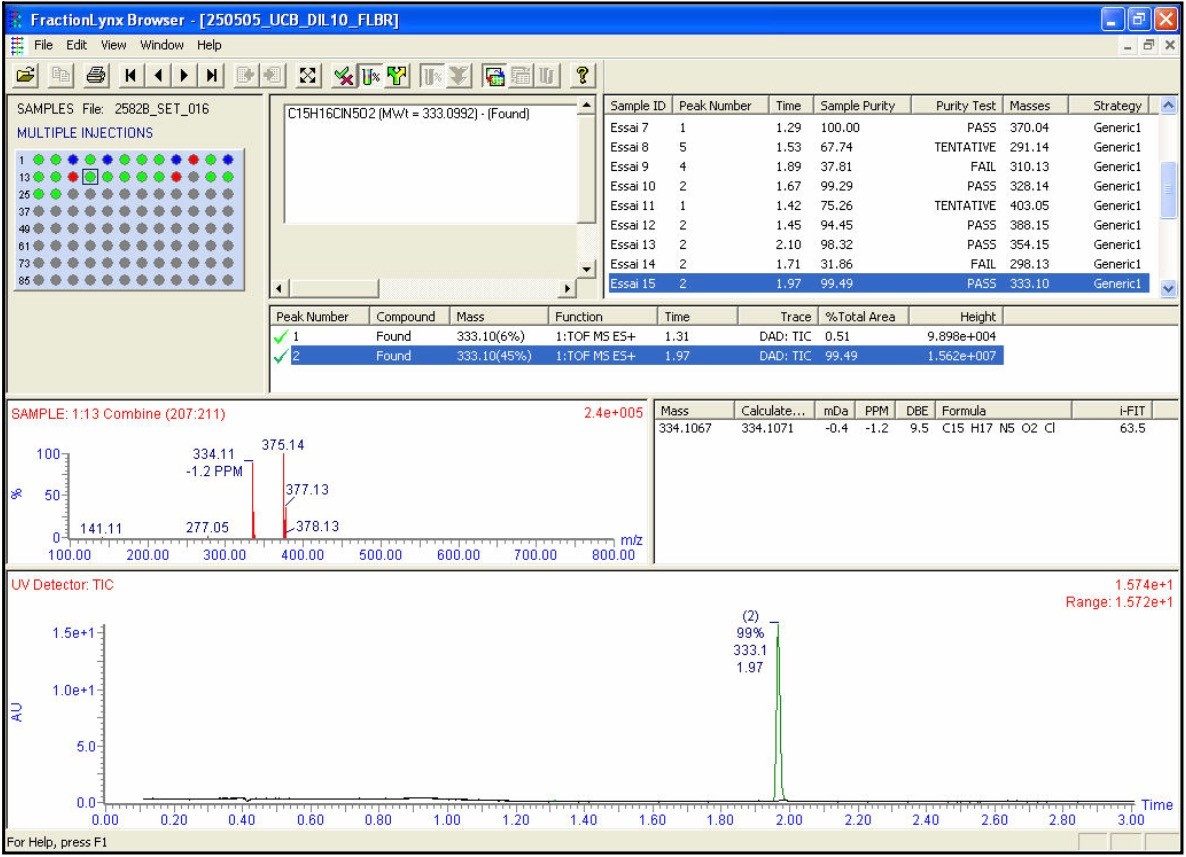
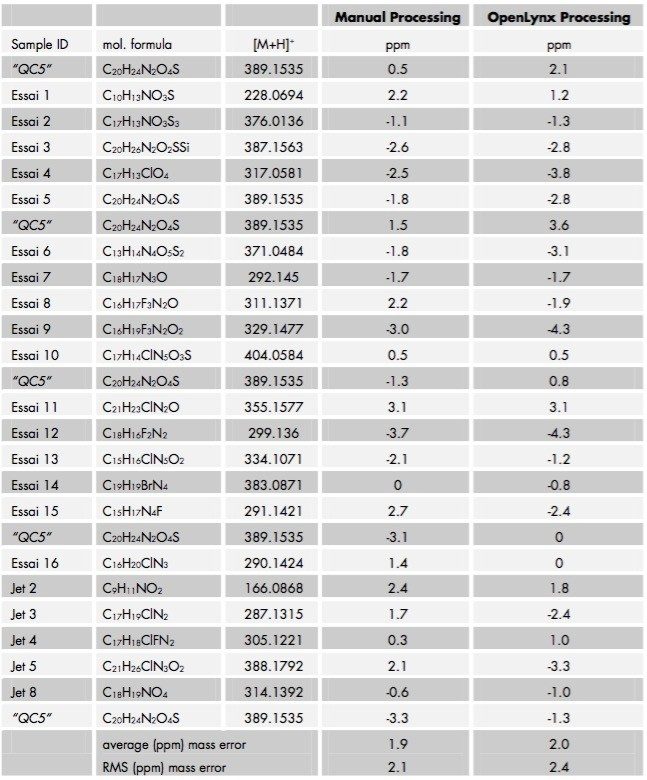
A fast, generic liquid chromatographic method at high pH has been designed to provide excellent selectivity for the investigation of basic compounds without compromise of either chromatographic resolution or speed of analysis. To obtain such an analytical method, Ultra Performance LC (UPLC) in conjunction with oa-ToF MS detection has been employed. With this analytical system, identification of the anticipated samples, isomers and possible impurities with mass accuracy deviations less than 5ppm from the actual were obtained using LockSprayTM. With such high accuracy data, the calculation of elemental compositions for each of the analytes was possible. Subsequent elemental composition results were produced using MassLynx i-FIT algorithm which takes into account the distribution of the spectral isotopes for the compounds of interest, and employs novel data interpretation to simplify results lists returned. To simplify and speed-up the processing of the sample batch, OpenLynx Application Manager was also utilized for fully automated QC of the compounds analyzed. Results were calculated for sample purity by UV, exact mass and elemental composition