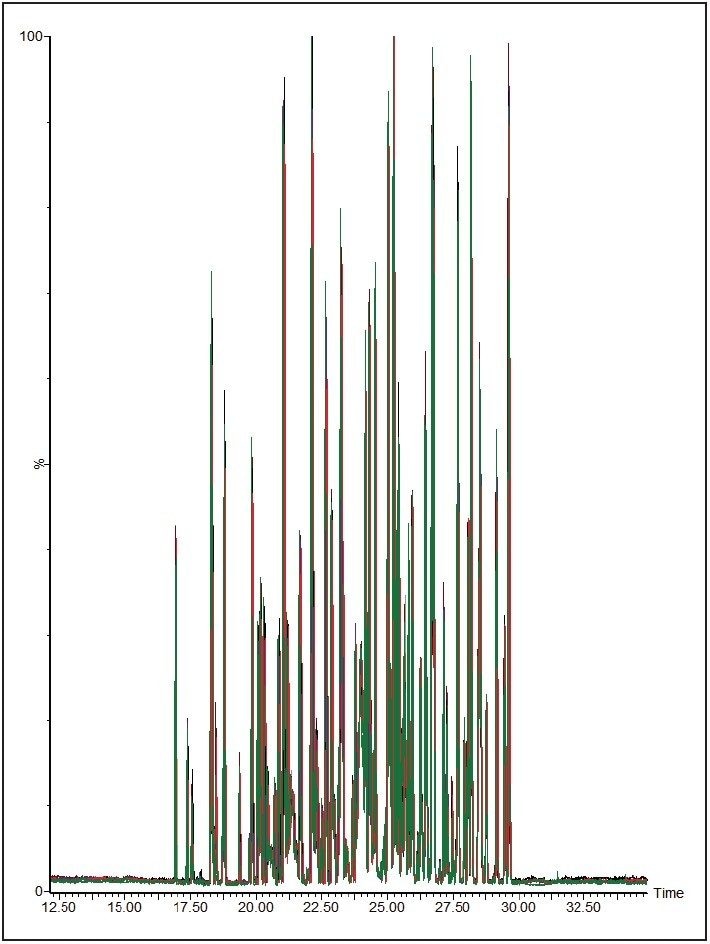
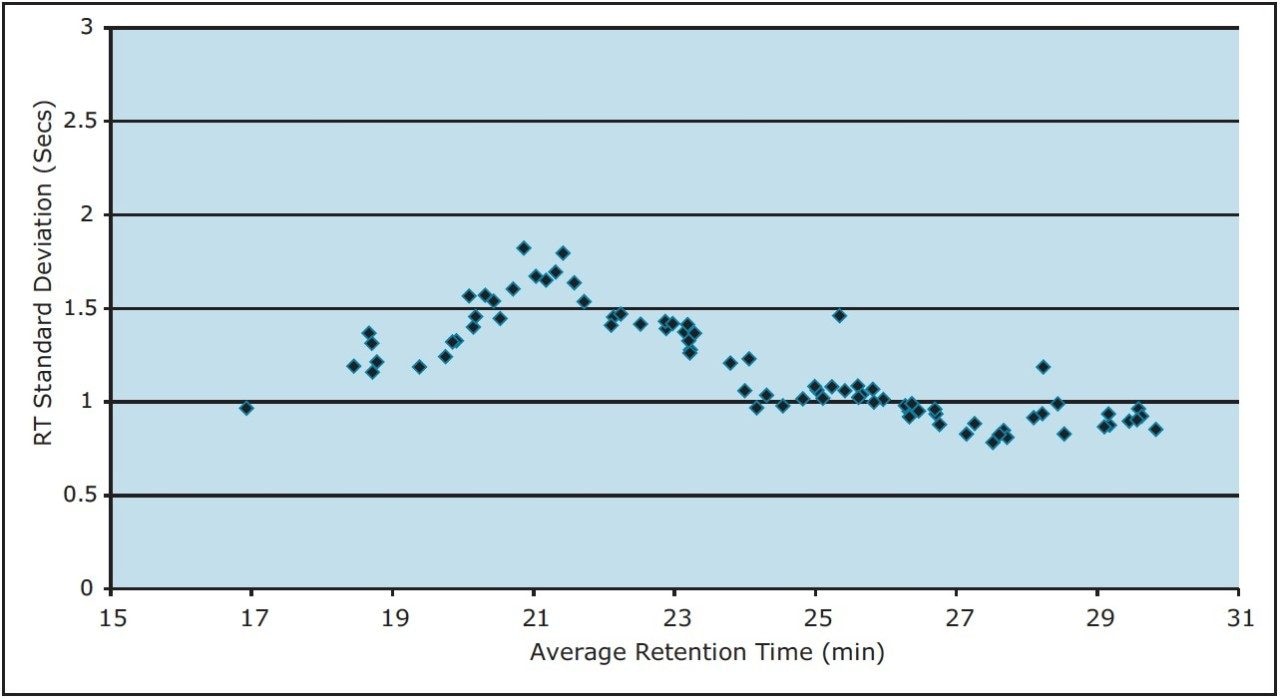
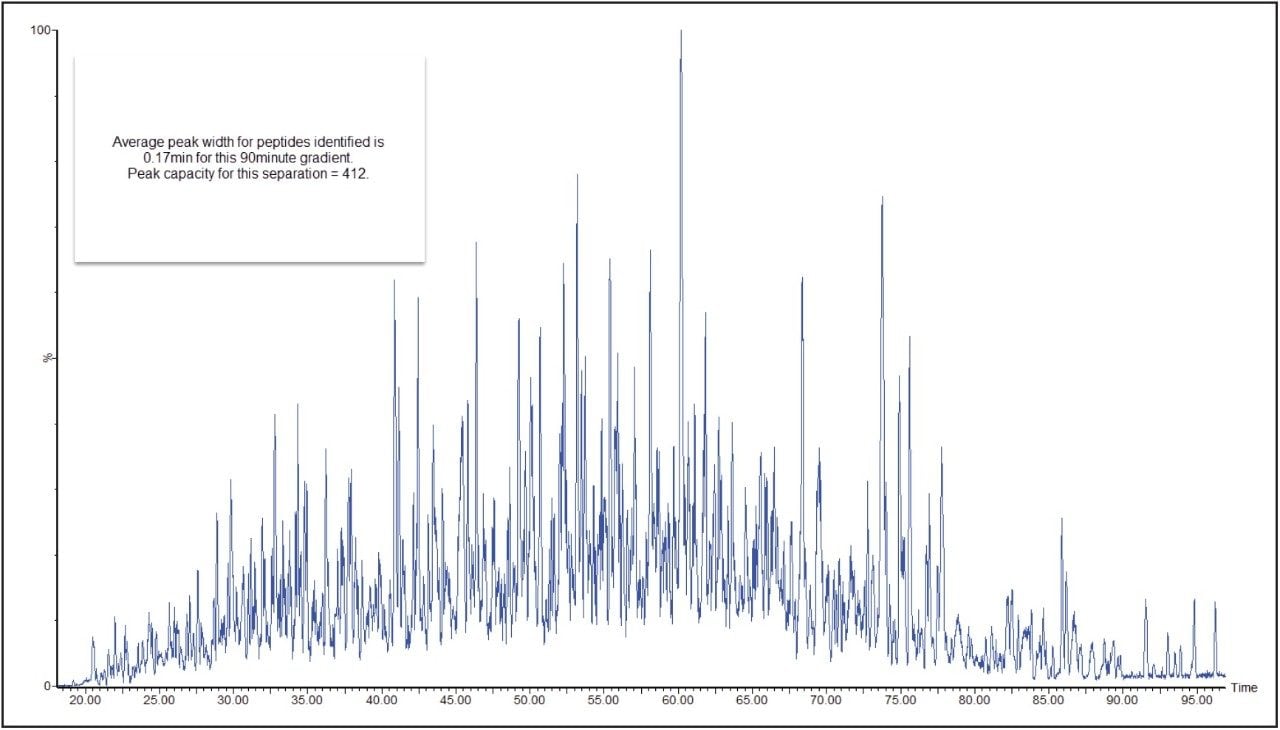
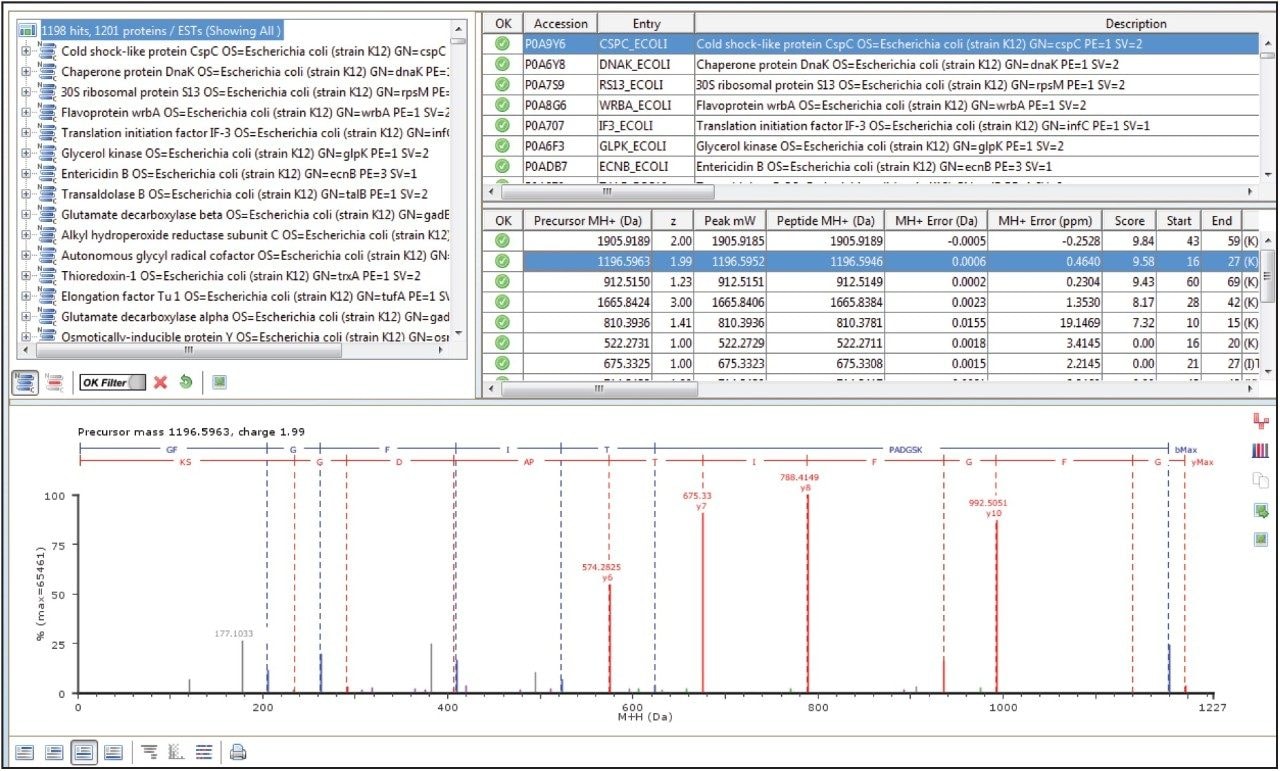
Nanoscale chromatography is established as the method of choice in bottom-up, or shotgun, proteomics experiments. It achieves superior sensitivity, compared with higher flow-rate separations, because of the reduced dilution effects of peptides of low stoichiometry present within the sample. When this scale of chromatography is coupled to a QToF mass spectrometer operating in the data-independent (DIA), data-dependent (DDA), or multiple-reaction-monitoring (MRM) acquisition mode, its higher peak-capacity separations yield enhanced results, namely, higher levels of protein/peptide identifications and lower detection and quantitation limits.
The ACQUITY UPLC M-Class System offers direct-flow separation for flow rates ranging from nanoscale to microscale, with an upper limit of 15,000 psi operating pressure. This higher pressure limit, compared with the nanoACQUITY UPLC System, permits the use of longer columns packed with sub-2-μm particles, for maximum separation efficiency. It also enables higher flow rates when shorter columns are used for high-throughput analyses.
This application note demonstrates the salient performance characteristics of the ACQUITY UPLC M-Class System, reporting typical results from DIA HDMSE acquisitions of a complex, tryptic-peptide sample.