Recent advances in mass spectrometry (MS) have enabled the simultaneous analysis of a wide range of chemically similar lipids as well as structurally diverse lipid classes, contributing to an increased interest in lipidomics research. However, the spatial localization of lipids within tissue micro-structures is often lost during the process of lipid extraction, as applied in more traditional analysis approaches, resulting in the loss of valuable information pertaining to origin and biological function.
Mass spectrometry imaging (MSI) visualizes the location of lipid species in entire tissue sections. The first step is typically an untargeted MS analysis experiment that enables large numbers of species to be detected and localized simultaneously. Structural identification of the detected lipid species is the next step; however, this can be time-consuming since it consists of manually conducting a series of MS/MS acquisitions on selected components, using either single or consecutive tissue sections.
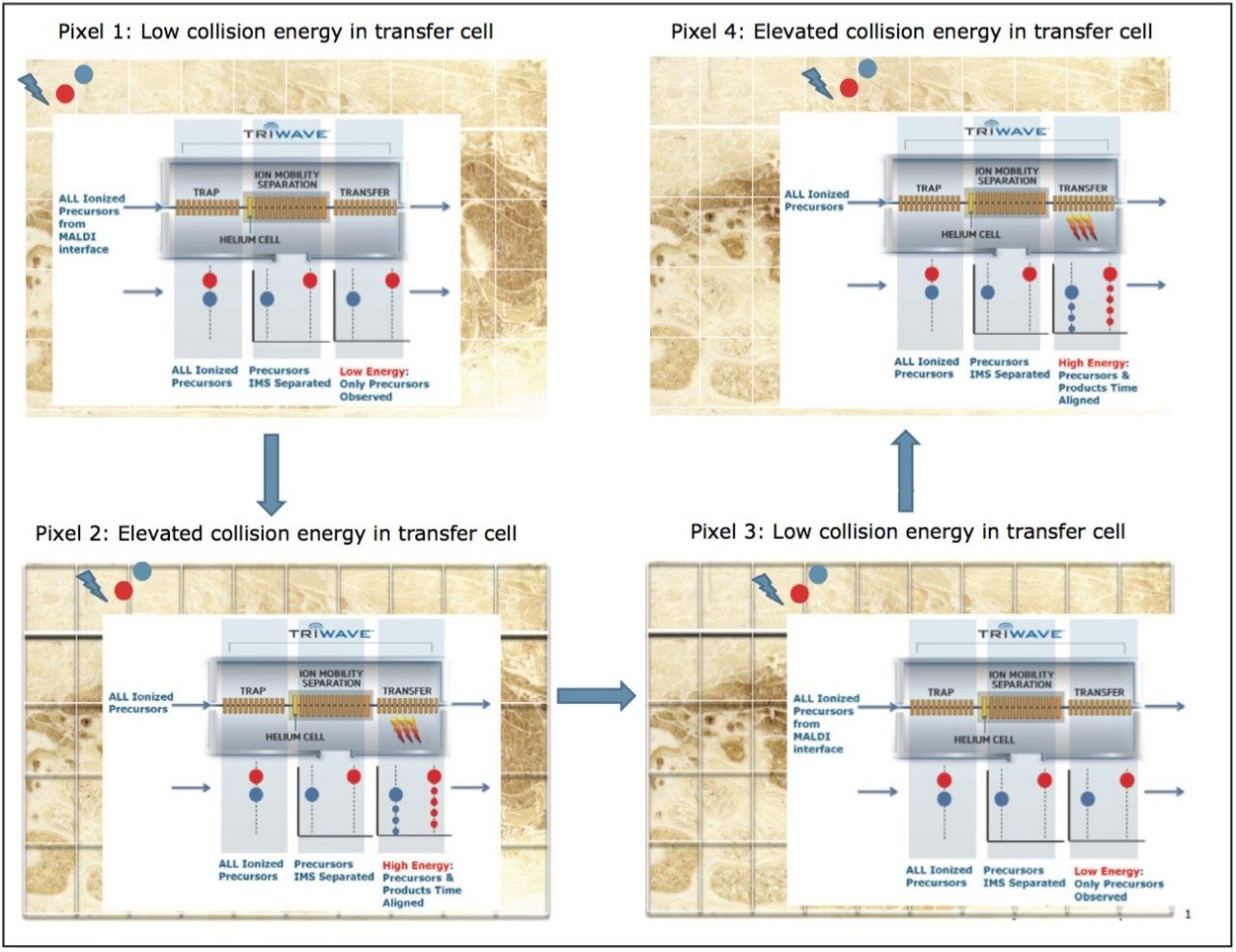
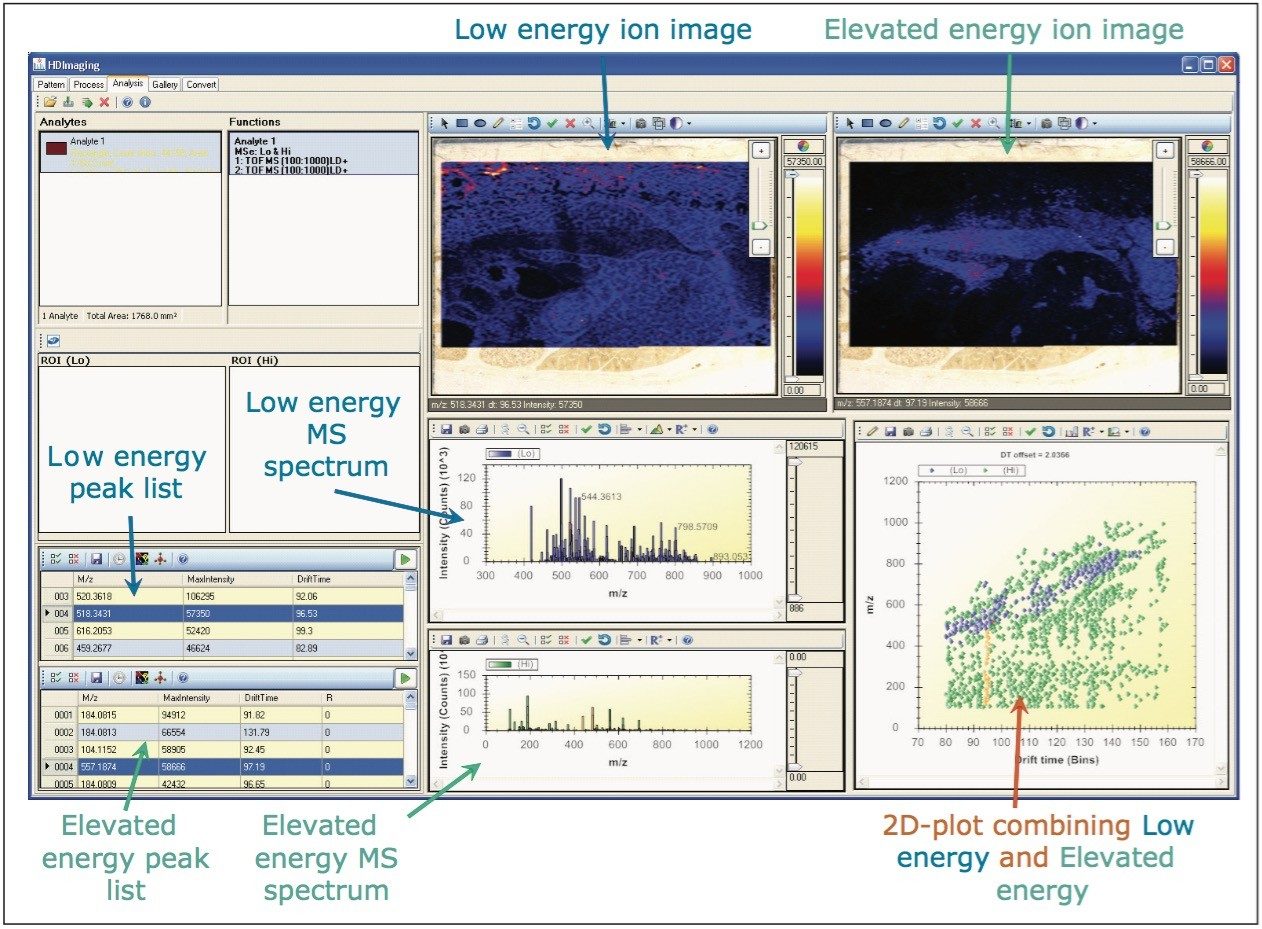
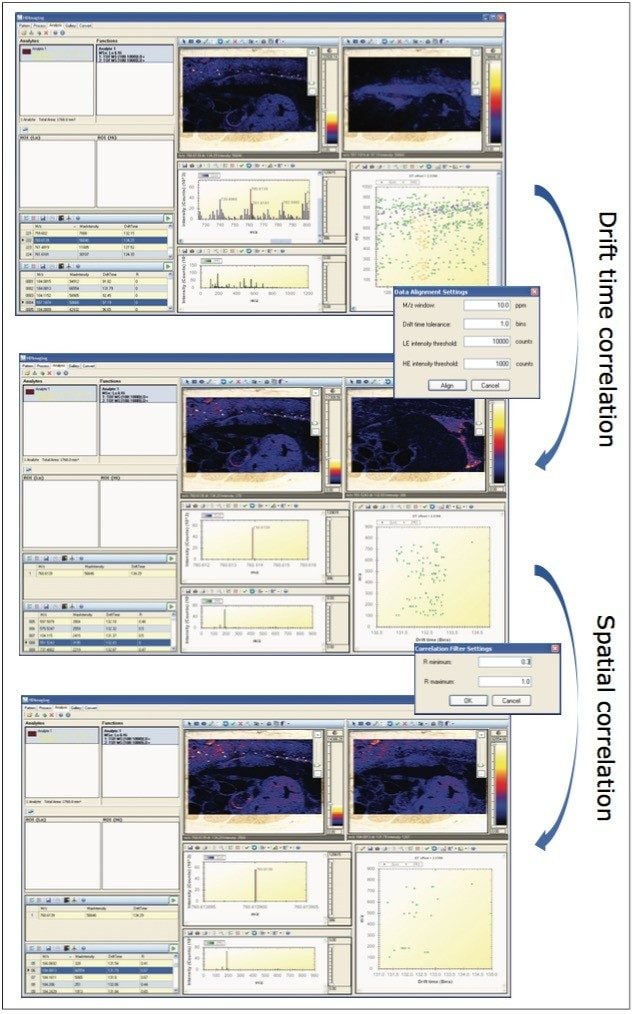
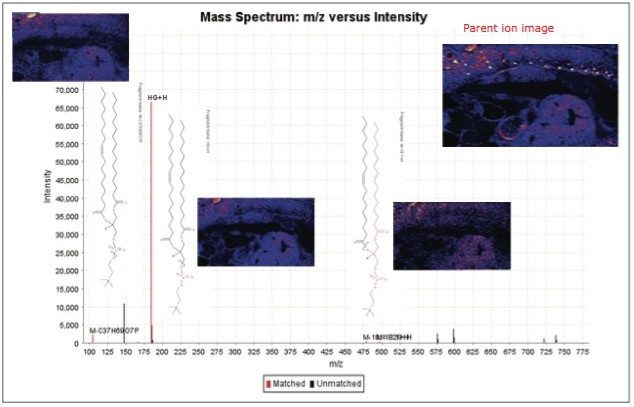
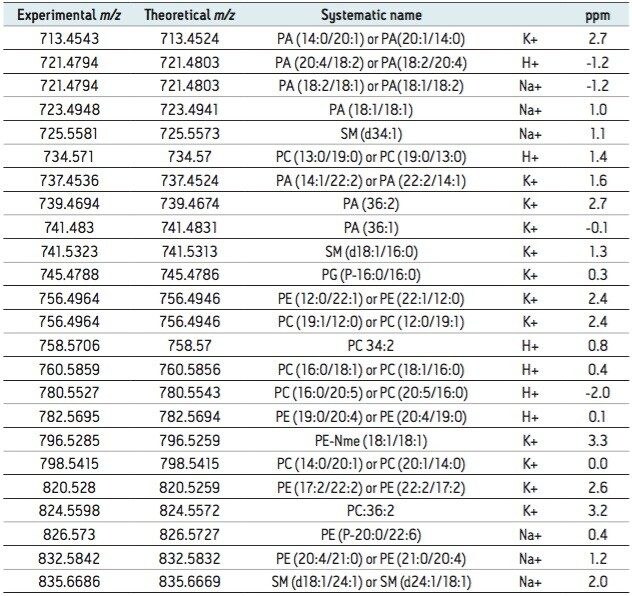
A data independent MALDI imaging acquisition method called MALDI Imaging High Definition MSE (HDMSE), presented here, enables detection and identification of lipid species in a single analytical run. This unique methodology provides MS and MS/MS information from detectable ion species within the same experiment, without the need for precursor selection. Post acquisition, precursors and fragments are correlated on the basis of ion mobility (drift time) and spatial distribution to provide highly informative results for every detectable molecular component.