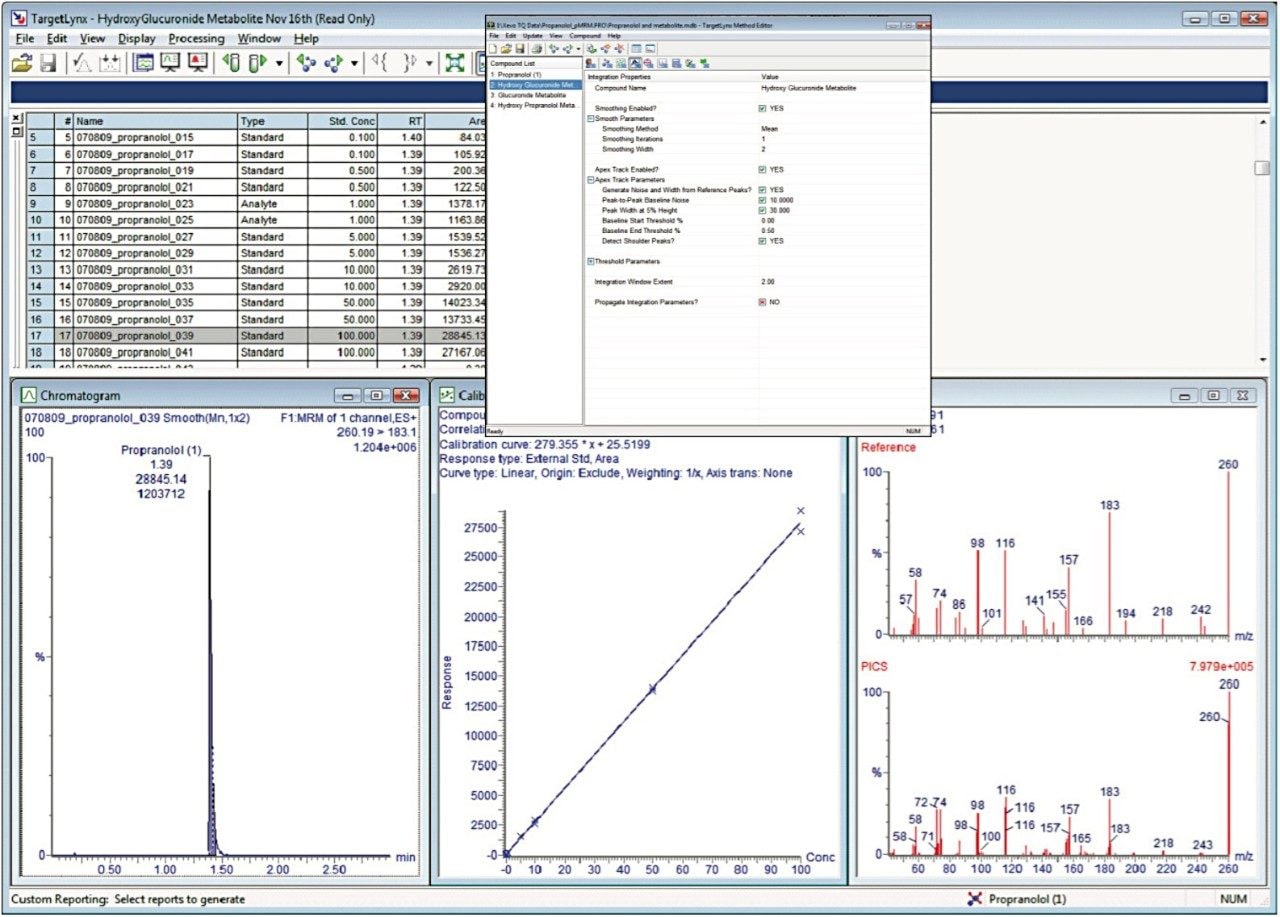
From the data it is possible to detect two hydroxy metabolites of propranolol (m/z 276), two glucuronide metabolites of propranolol (m/z 436), and three hydroxyglucuronide metabolites of propranolol (m/z 452). This data is in agreement with that of Wilson, et al,3 who detected the same number of hydroxy, glucuronide, and hydroxyglucuronide metabolites. The MRM peak area data showed a linear response relative to concentration over the range of 10 pg/mL to 50 ng/mL (without the use of an internal standard, data not shown).
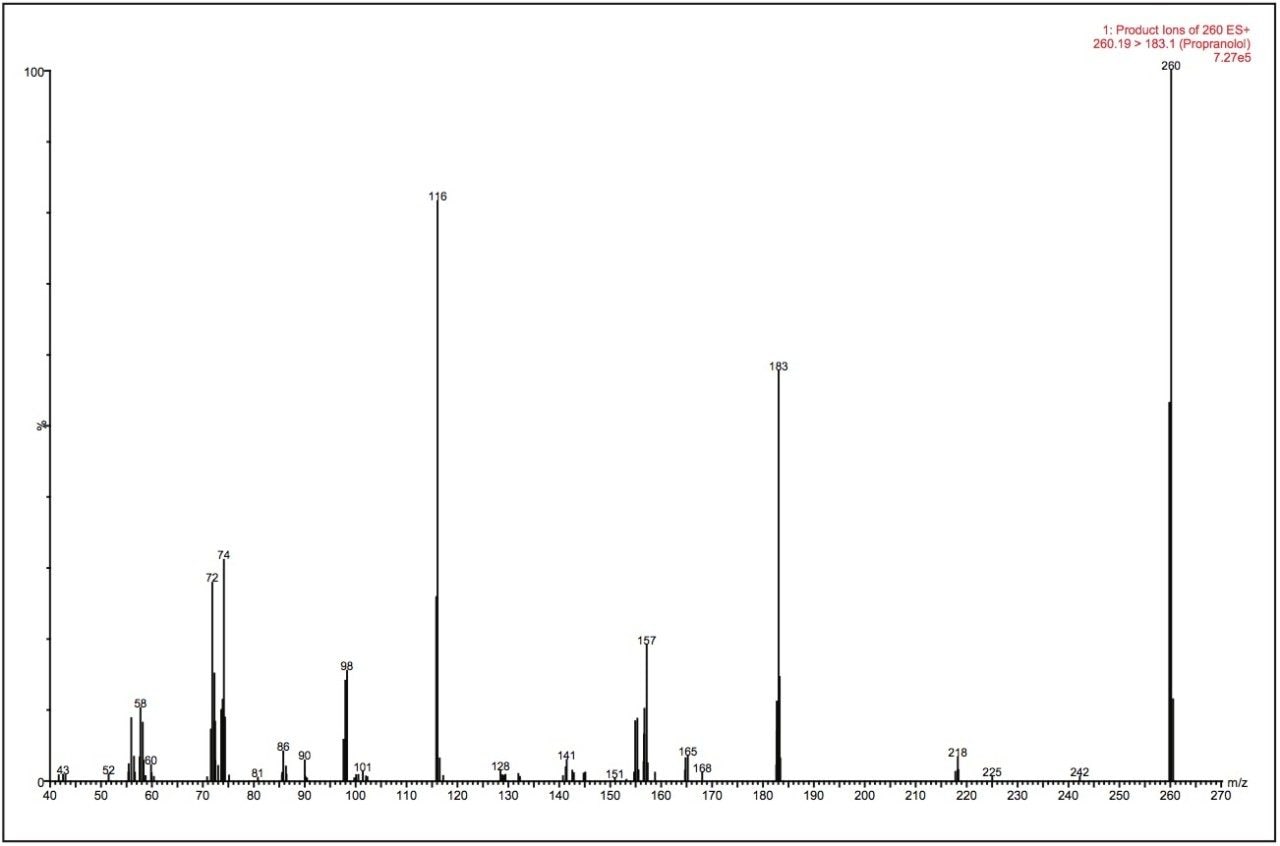
The extracted ion chromatograms and mass spectra produced are displayed in Figure 3. The LC-MS/MS system detected three hydroxyglucuronide metabolites, two glucuronide metabolites, and one hydroxy metabolite of propranolol eluting with retention times of 0.76, 0.86, and 1.13 minutes, respectively. Representative spectra of the hydroxyglucuronide (RT 0.86 min) are shown in Figure 3A. The spectra show the major ions m/z 276, 199, and 116. These correspond to the hydroxylated phase I metabolite and the fragment ion associated with this metabolite. The m/z 116 ion is a common fragment ion from the parent molecule. The mass spectrum displayed in Figure 3B is from the glucuronide metabolite eluting with a retention time of 1.33 minutes.
In the spectra shown in Figure 3, we can clearly see the m/z 260 ion produced from the cleavage of the sugar moiety from the propranolol analyte. The m/z 183 and 116 ions come from the fragmentation of the propranolol molecule itself. The data displayed in Figure 3C show that there are two peaks detected. A careful examination of the PIC spectra showed that the peak with a retention time of 1.13 minutes is actually from the thermal degradation of one of the hydroxylated glucuronide metabolites in the MS ion source and not a second hydroxylated metabolite. The PIC MS/MS spectra of the second, smaller peak at 1.24 minutes gave rise to peaks with m/z values of 116, 161, 198, and 233. The 116 ion is common to all metabolites. The 161 ion comes from the oxidation of the 145 ion, and the 198 ion comes from the addition of 16 to the 182 ion.