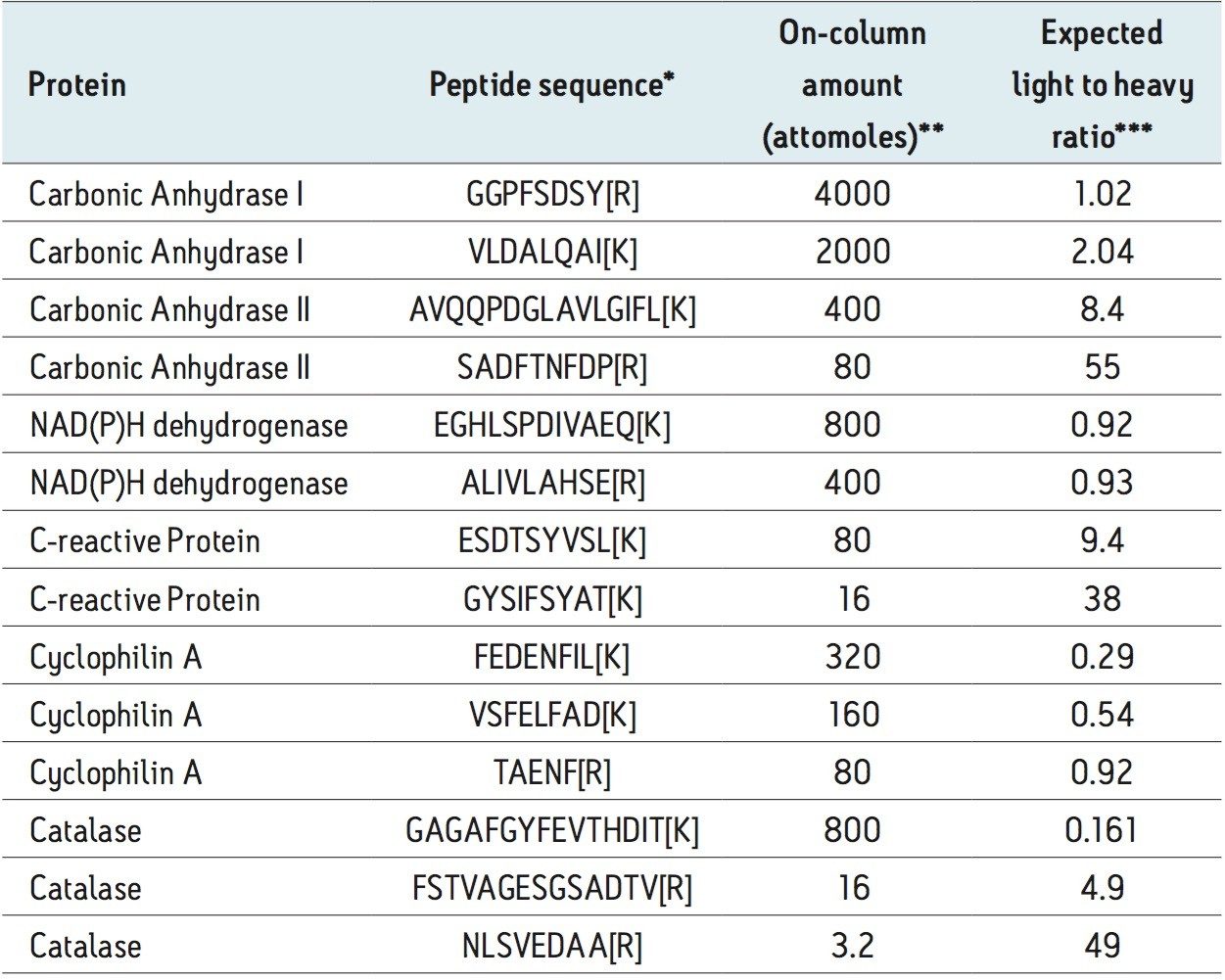
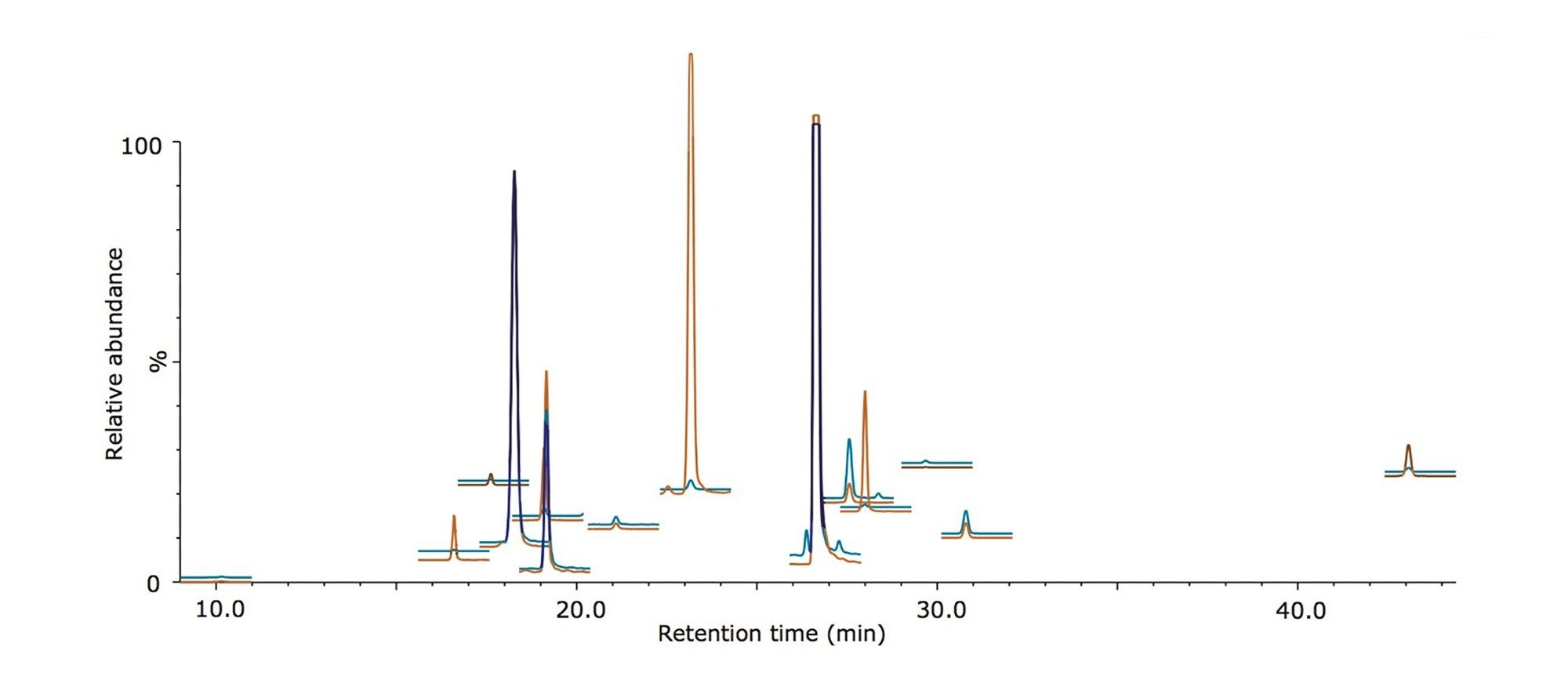
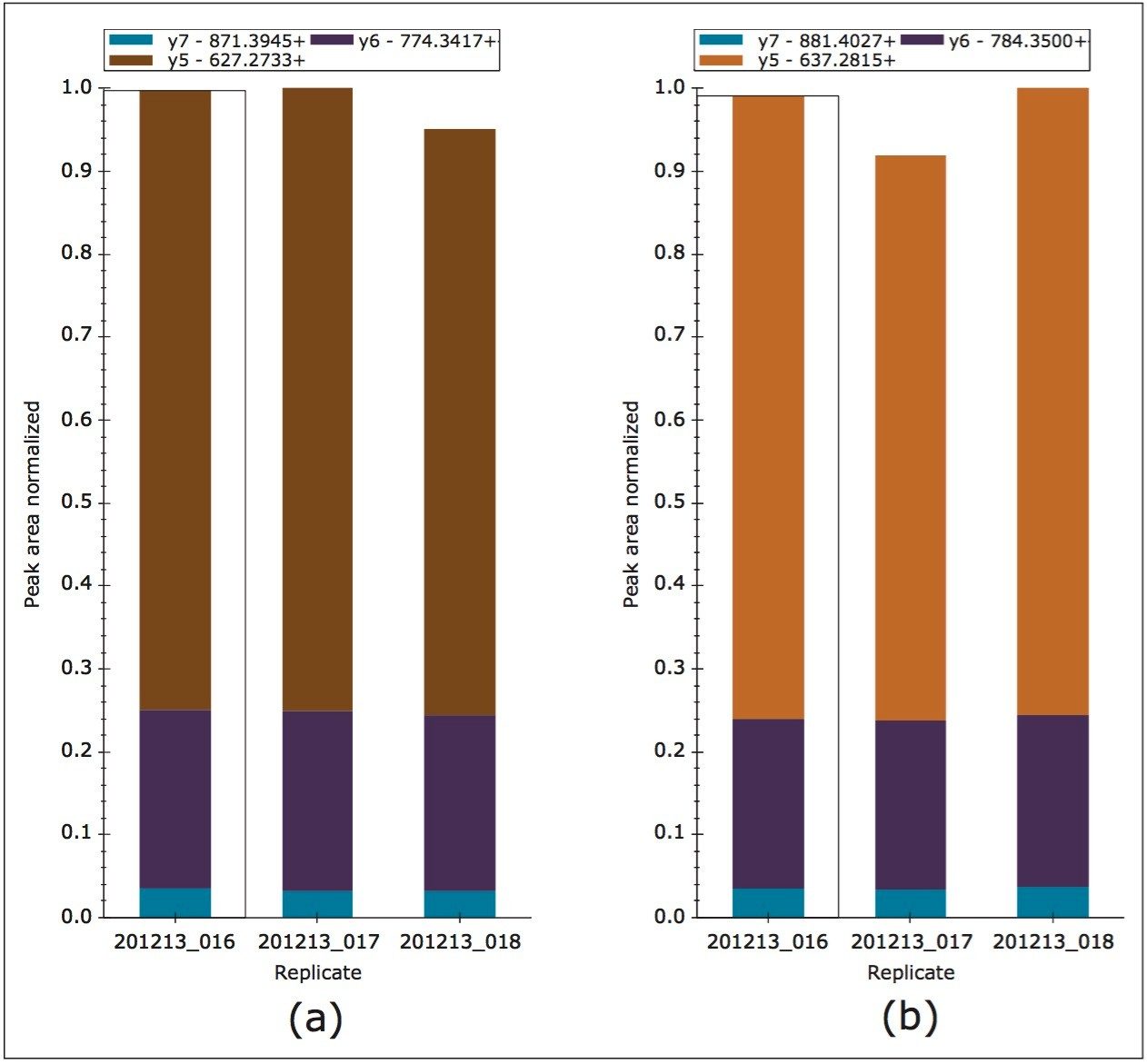
Biomarker discovery and validation are the first two steps in understanding disease and drug development. Validation is technology-challenged since it requires analyzing a large number of samples with high-throughput, but nevertheless requires high sensitivity, high resolution, large dynamic range and excellent selectivity.
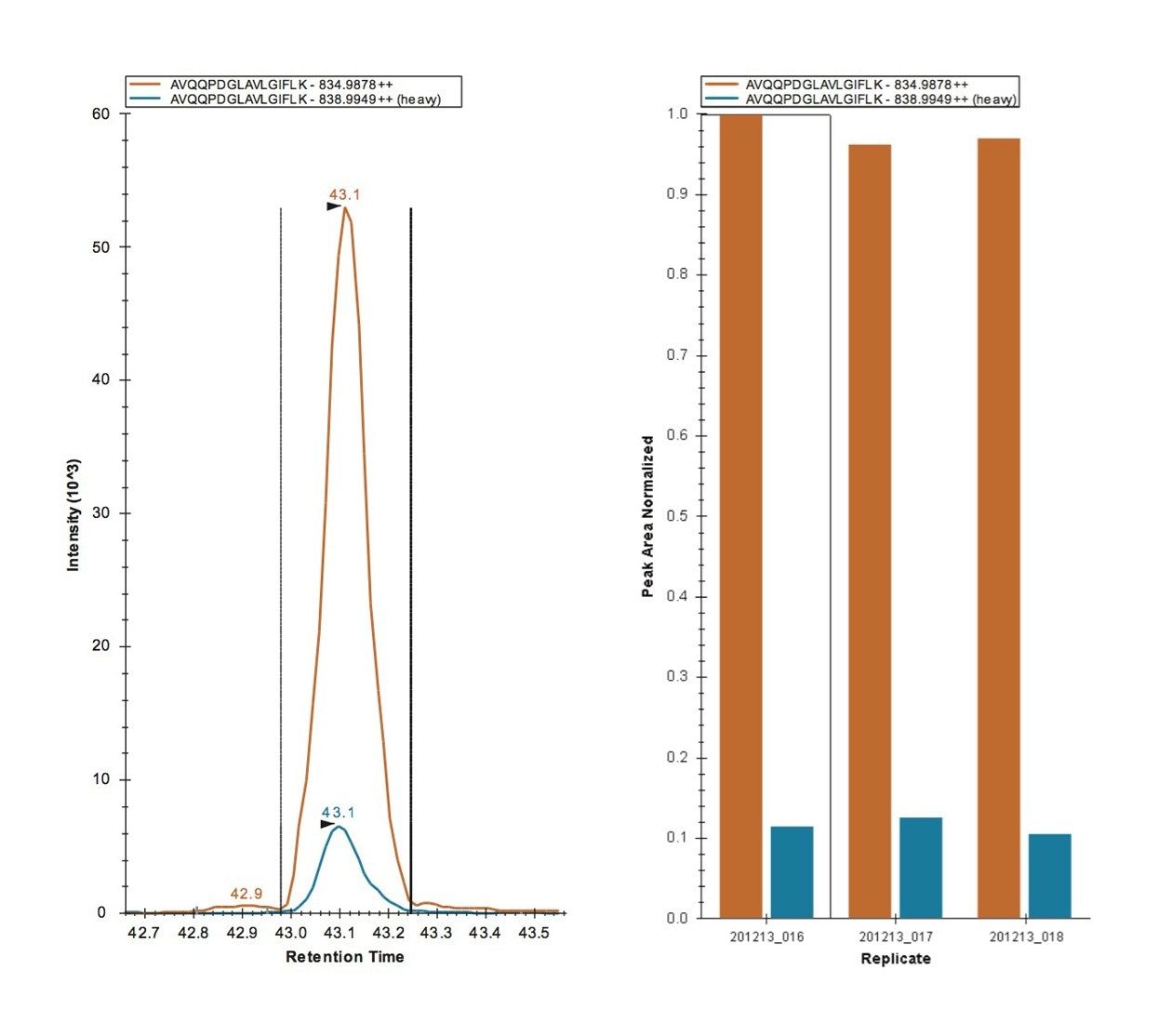
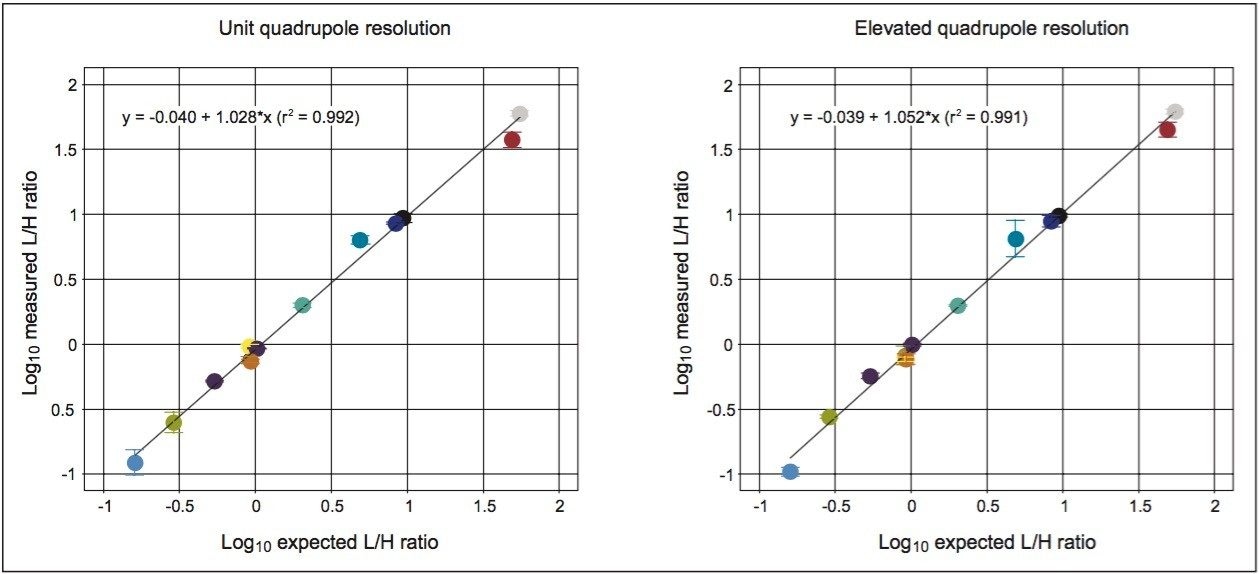
Targeted LC-MS based assays afford protein quantification with the reproducibility and throughput required to improve marker acceptance. Multiple reaction monitoring (MRM) using tandem quadrupole mass spectrometers is one of the enabling technologies applied in targeted LC-MS approaches.1 Overall, MRM-based methods compare favorably with antibody-based techniques, such as ELISAs or protein arrays, in that MRM-based methods are less expensive and can be developed more rapidly. However, one of the major challenges using MRM for candidate marker verification in mammalian body fluids is the required sensitivity for the quantification of low-abundance proteins, especially in the case of sample limited conditions.
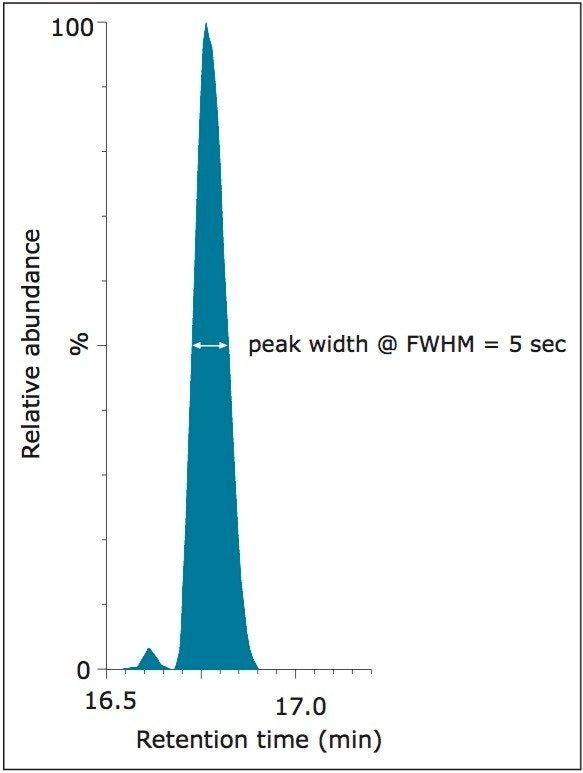
Miniaturized LC systems offer improved mass sensitivity but often lack the required throughput, robustness, and reproducibility. The application of a novel microfluidics platform for the quantification of marker peptides and proteins is presented, considering speed, sensitivity, and selectivity.