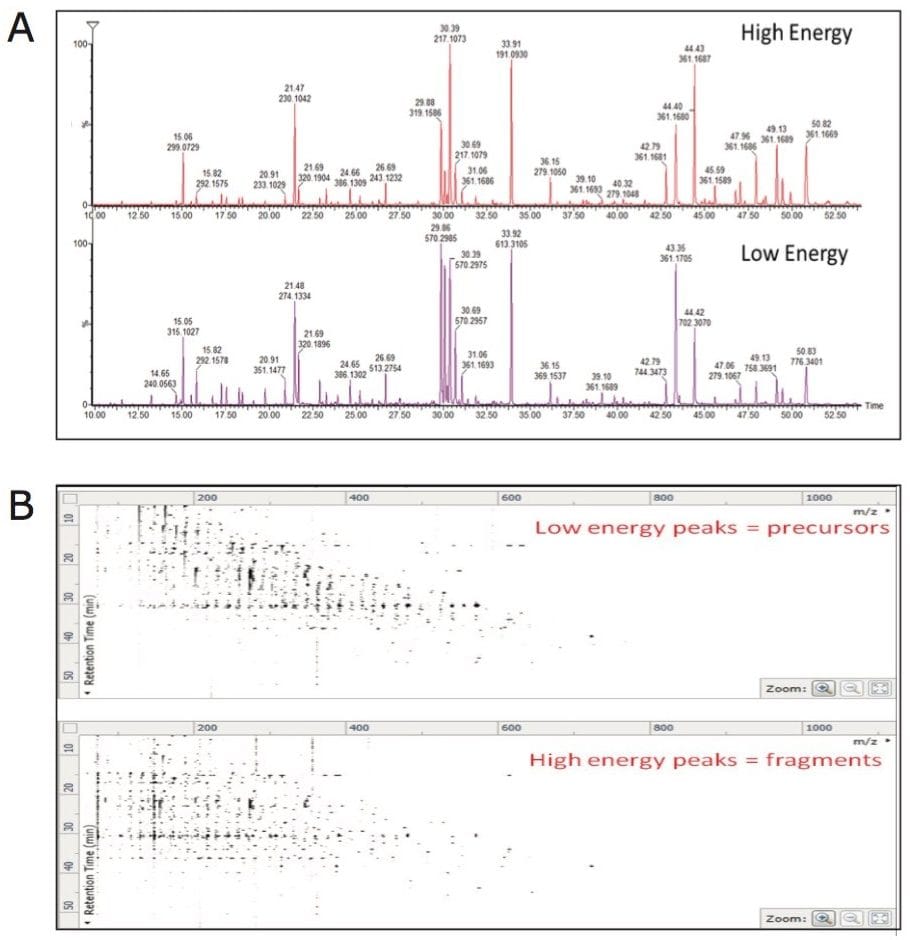
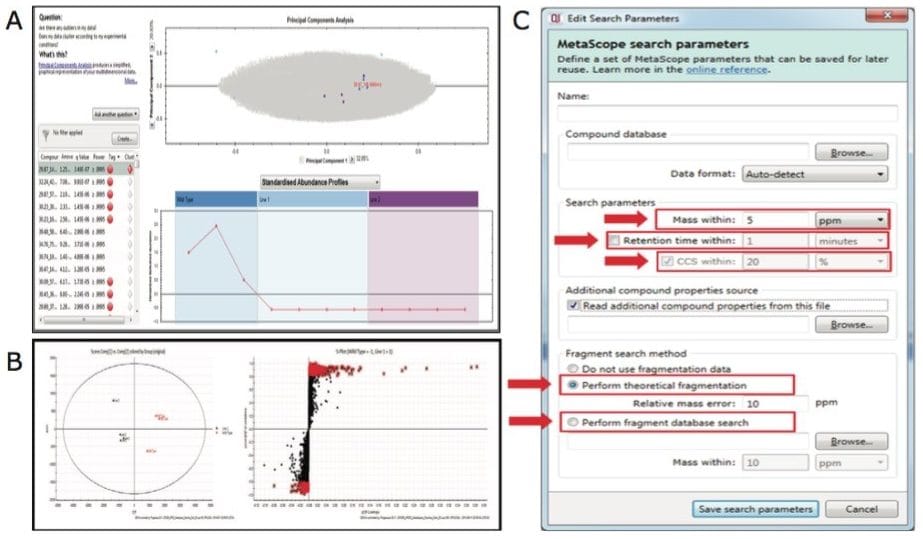
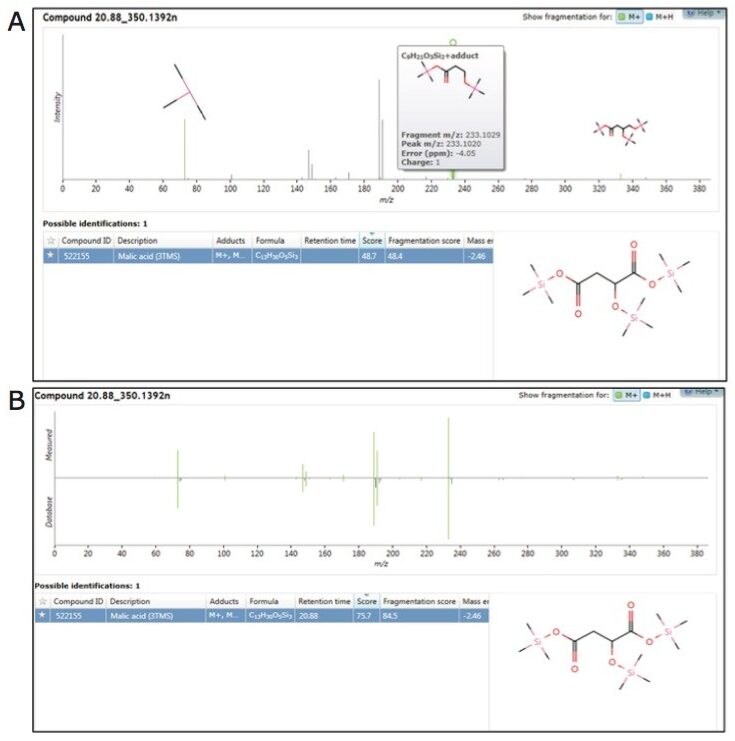
Waters offers a comprehensive range of analytical system solutions, software, and services for scientists. Liquid Chromatography. Mass Spectrometry. Waters is the leading provider of lab equipment, supplies and software for scientists across the world. Easily research and order everything your lab needs! Waters offers a comprehensive range of analytical system solutions, software, and services for scientists. Liquid Chromatography. Mass Spectrometry. Waters is the leading provider of lab equipment, supplies and software for scientists across the world. Easily research and order everything your lab needs!

![Figure 1. A) An APGC System can be coupled to various Waters MS instruments, including the Xevo TQ-S and the SYNAPT G2-S. The changeover from UPLC to APGC takes less than five minutes. B) The APGC source consists of an ion chamber with a corona pin inside. The GC column enters the source via a heated transfer line. Corona discharge creates nitrogen plasma within this region. Radical cations generated in this plasma interact and ionize the analyte molecules. The ions created are then transferred to the mass analyzer. C) Under dry source conditions the predominant method of ionization is charge transfer, generating molecular radical cations [M+•]. This method favours relatively non-polar molecules. D) When a protic solvent, such as water or methanol, is added to the source, the predominant ionisation is proton transfer, generating [M+H]+ ions. This method favors relatively polar molecules.](/content/dam/waters/en/app-notes/2015/720005298/720005298en-f1.jpg.82.15-14-683-969C.resize/img.jpg)