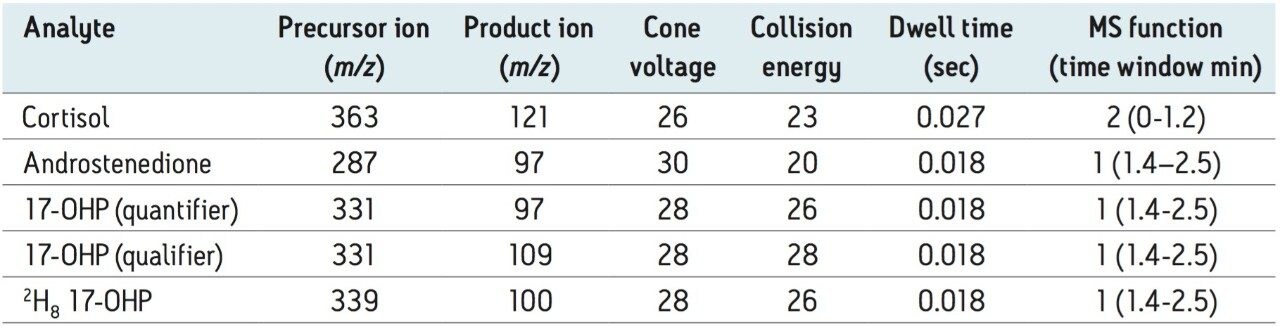
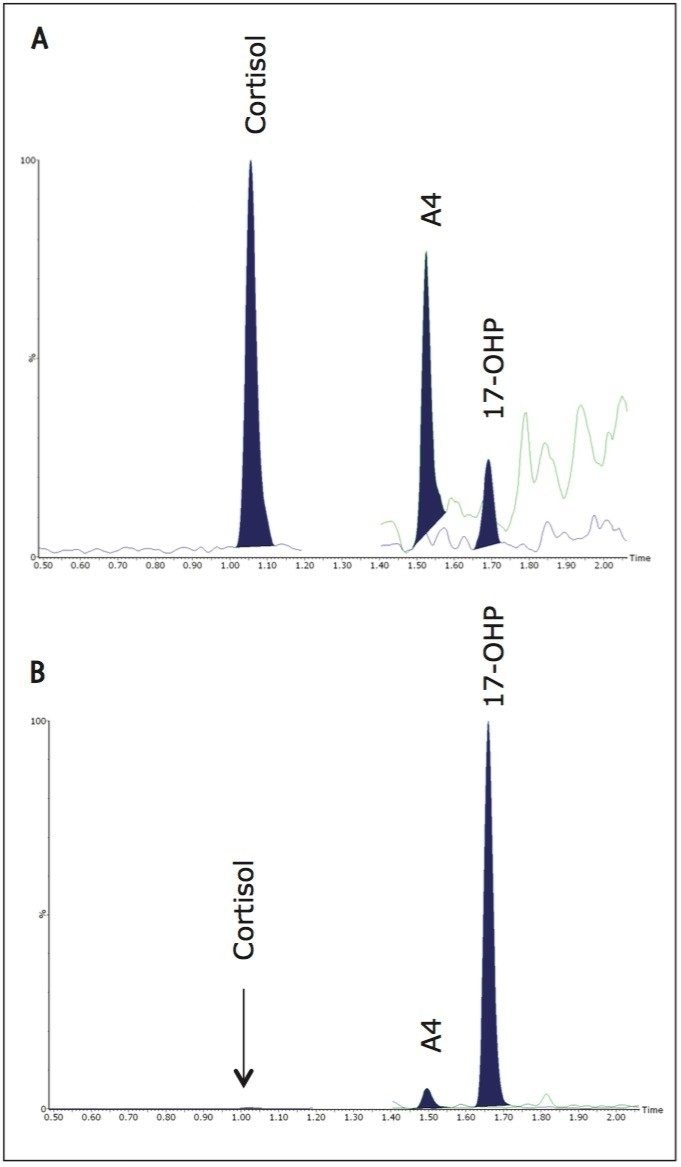
Within- and between-batch imprecision, expressed as coefficient of variation of replicate measurements of independently-prepared quality control DBS at target mean 17-OHP concentrations 76, 151, and 303 nmol/L was < 6.7% CV (between-batch in singlicate over 5 days; within-batch n=5). The between-batch imprecision of the lowest calibrator 9.9 nmol/L was 7.3% CV (singlicate over 5 batches).
An artificial whole blood matrix was prepared for evaluation of extraction efficiency. The negative control blood matrix along with 3 positive controls of blood matrix spiked to 76, 152, and 303 nmol/L 17-OHP were spotted onto Whatman 903 filter paper. The dried residue of the extracted negative pool was resuspended in solvent standards at the maximum expected 17-OHP concentration for each level of the positive control extract, assuming total recovery of the spiked 17-OHP. The mean extraction efficiency of 17-OHP, calculated as the ratio of the peak area of the positive control: negative control post-extraction spiked sample, was 58% (57 – 59%, n = 3).
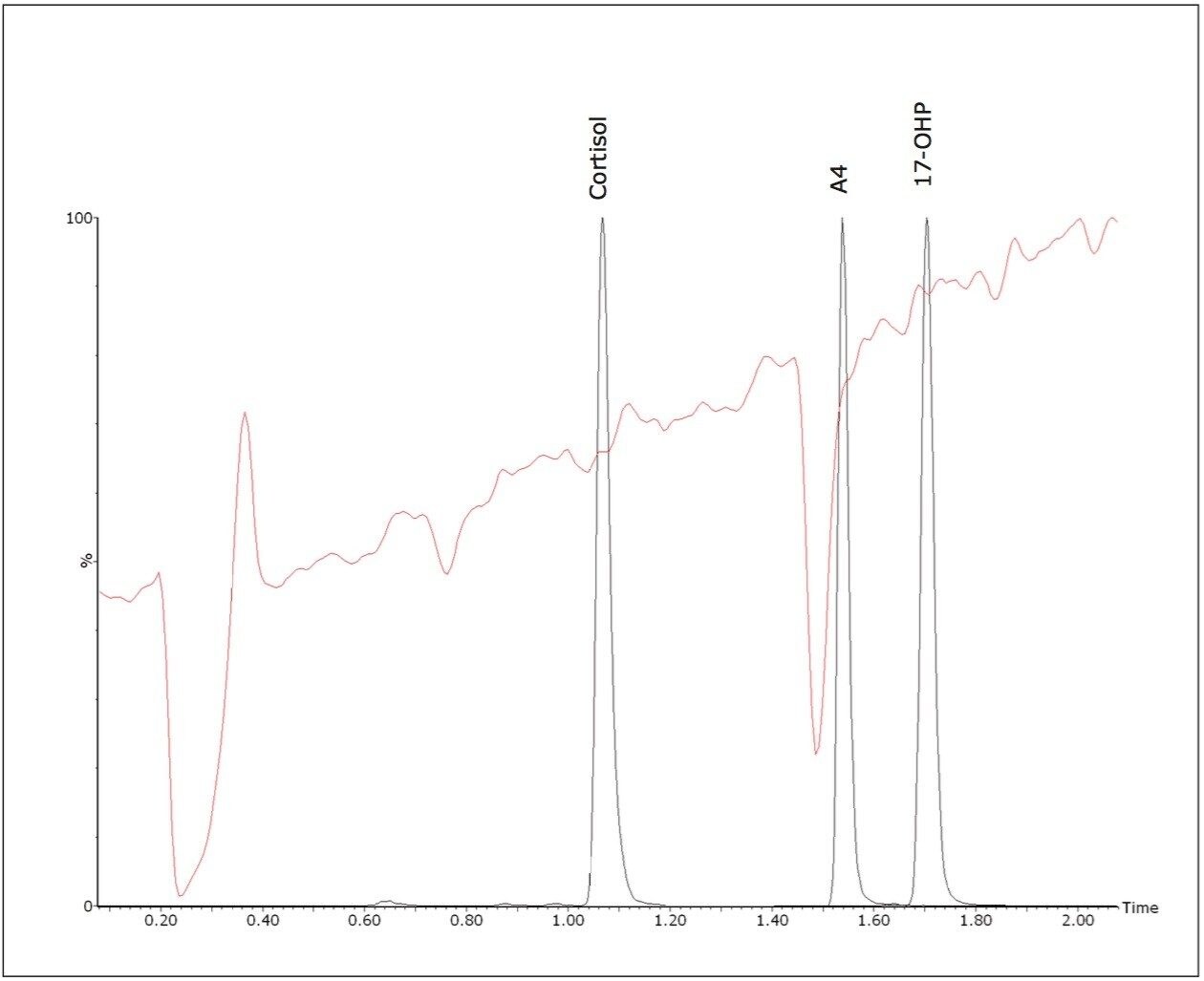
Quantitative matrix effects were assessed, expressed as the proportion of detector response suppressed or enhanced by the presence of matrix. The ratio was calculated of the peak area of post extraction spiked negative pool: peak area of matrix-free solvent standards of equivalent concentration. The mean signal suppression due to matrix was 15% (13 – 16%, n = 3). A qualitative evaluation of signal suppression conducted by post-column infusion of a solvent standard of 17-OHP into the LC flow path of extracted samples confirmed compounds of interest do not elute within regions of signal suppression (Figure 3).