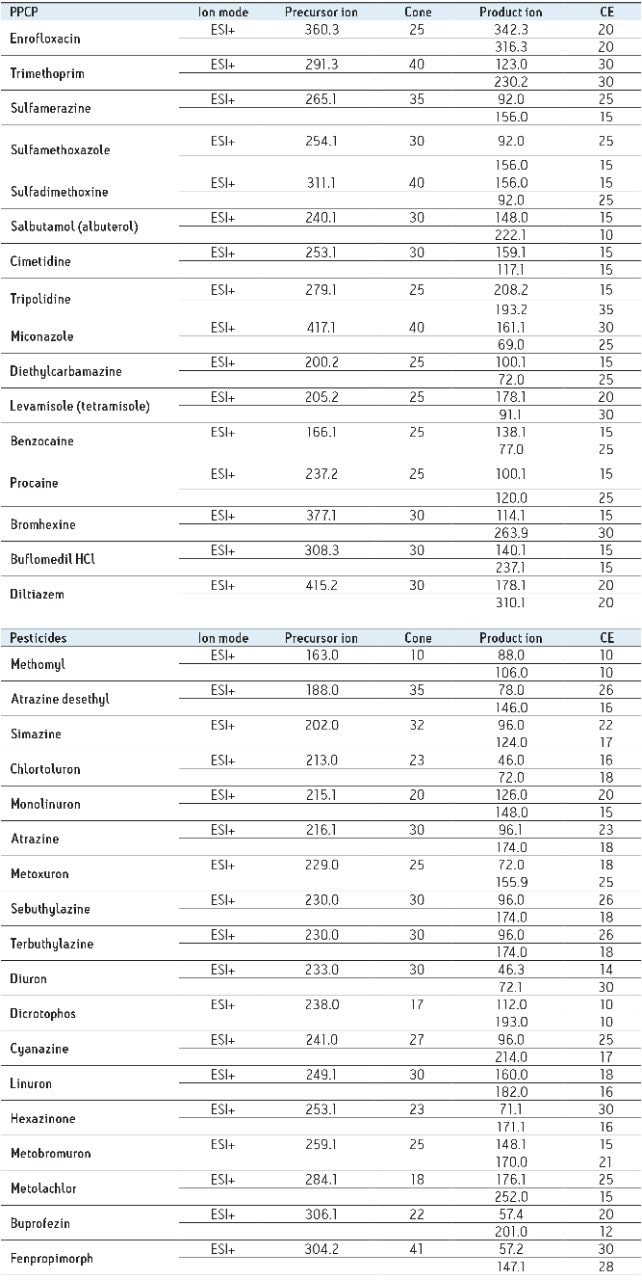
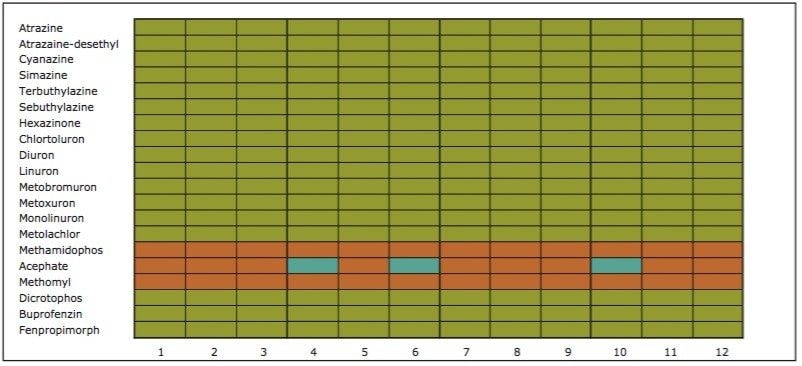
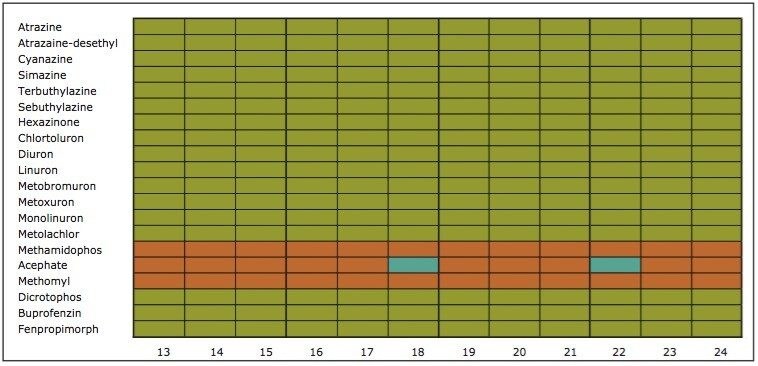
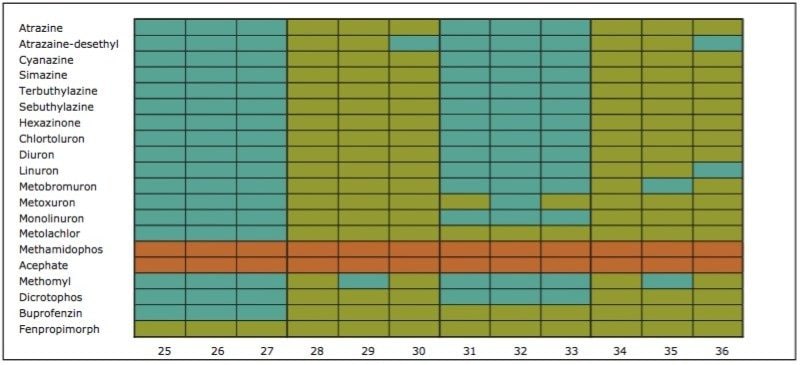
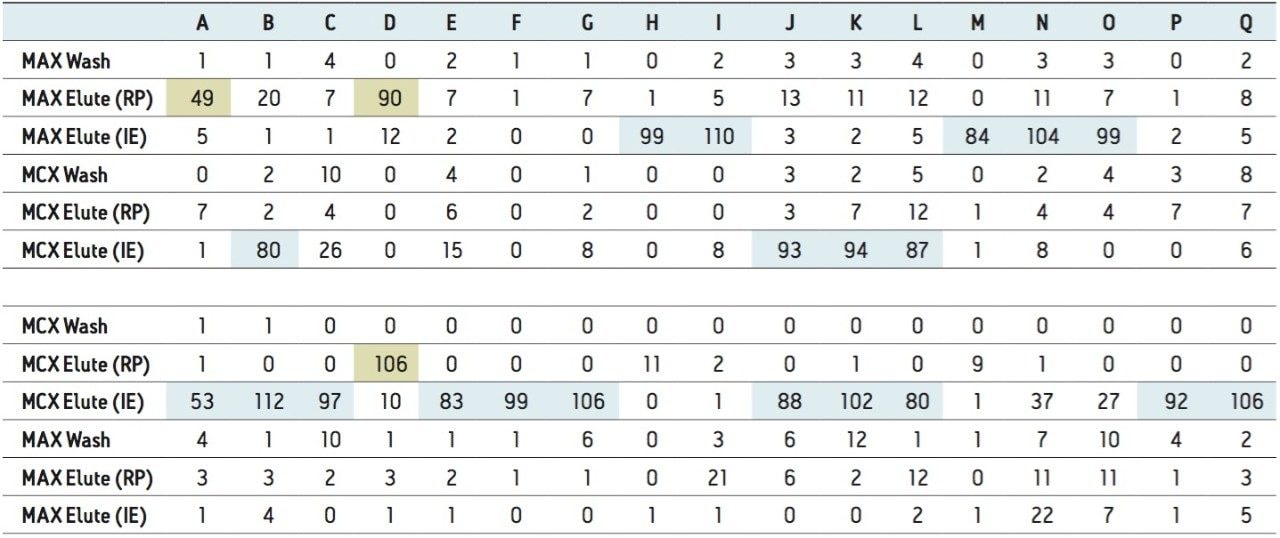
The extraction sequence starts with a sorbent conditioning step to remove potential interferences. The next step is sample loading, which extracts target analytes from the sample. Typical loading flow rate for large sample size range is between 5 mL/min and 10 mL/min. The loading flow rate is an optimized function derived from the SPE bed mass, sample contact time, and mass transfer onto the sorbent. With a loading flow rate set at 10 mL/min, the total loading time should take 1.6 hours before proceeding with the next step of the extraction protocol. However, as seen in Figure 1, the values for high-grade water (2.5 hours), tap water (8 hours), and surface water (15 hours) samples far exceed the expected 1.6 hours. The discrepancy comes from the fact that the loading flow rate is not at a constant value for the entire sample volume. In fact, the flow rate is linked to the quality of the sample and, therefore, the extended loading time is attributed to clogging issue from particulate matter in the sample. This is necessary to reach the desired target LOD or LOQ. In some instances, it may be necessary to extract a larger sample volume to increase the enrichment factor. Once the total volume is extracted, a wash step removes weak interferences without causing breakthrough for the target analyte. The elution step breaks the retention bonds of the target analyte from the SPE sorbent. At this point in the extraction process, the target analyte sustained a solvent exchange from aqueous to an organic solvent (aqueous or non-aqueous miscible). If the final extract is dissolved in a non-aqueous miscible solvent, this indicates that the analysis will be performed with a GC-MS platform. If the analysis is performed with an LC-MS and assuming a reversed-phase separation, the final extract must undergo a second solvent exchange. This is accomplished by using nitrogen stream evaporation to evaporate the sample to dryness and reconstitute with initial mobile phase conditions.
Nitrogen evaporation is linked to the properties of the organic solvent and any remaining percentage of water collected during the elution step. In some cases, the evaporation time can be decreased by applying mild heat. It is a well-known fact that evaporative loss is always a potential cause for poor recoveries. In some instances, the evaporation rate can be at extreme low settings, which requires adding an overnight time period for completion. Finally, once the sample is reduced to dryness, yet another cause of poor performance can occur by reconstitution solvent compatibility and solubility. The overall workflow is dependent of the analytical technique used for analysis and can be extremely time-consuming and laborious.
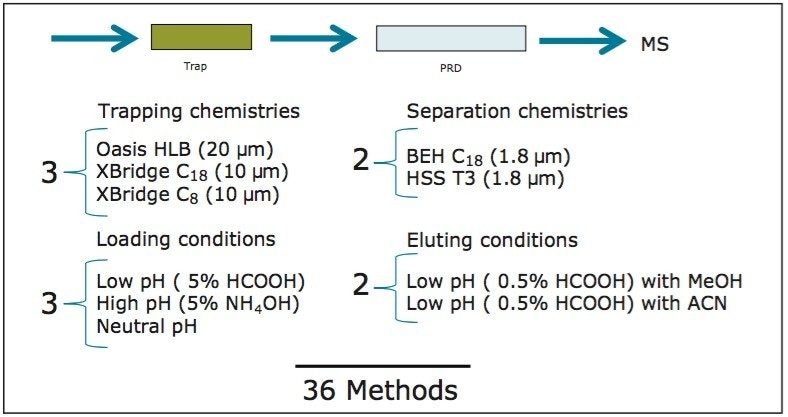
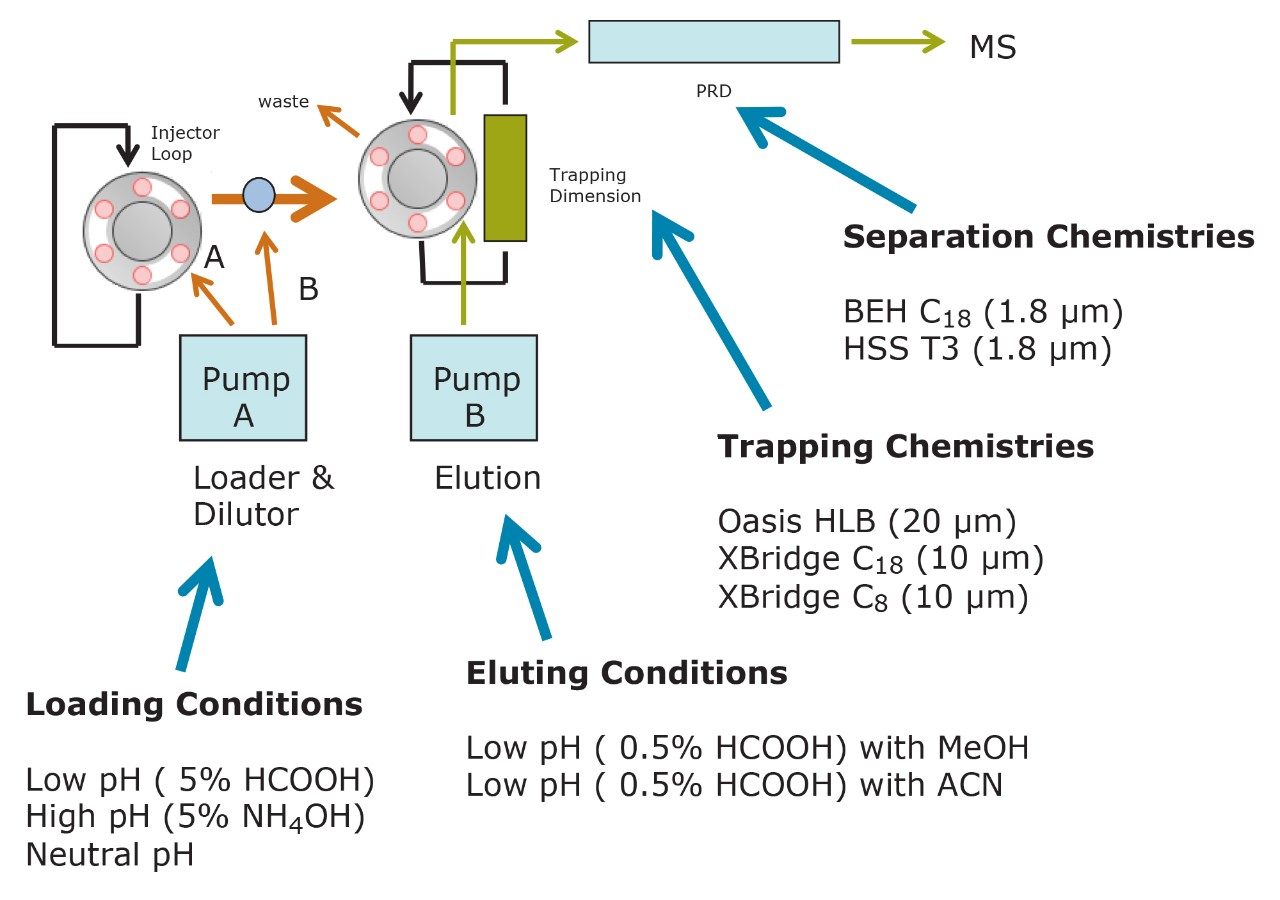
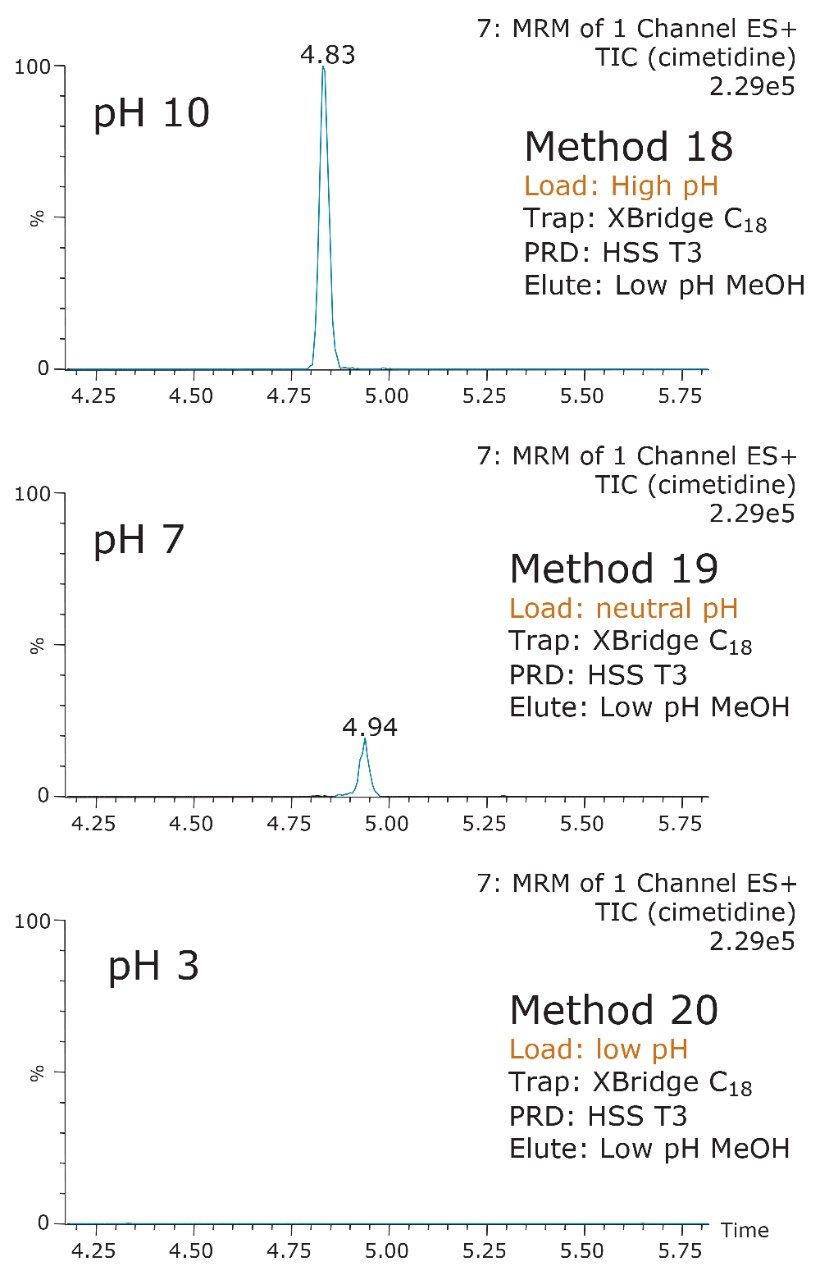
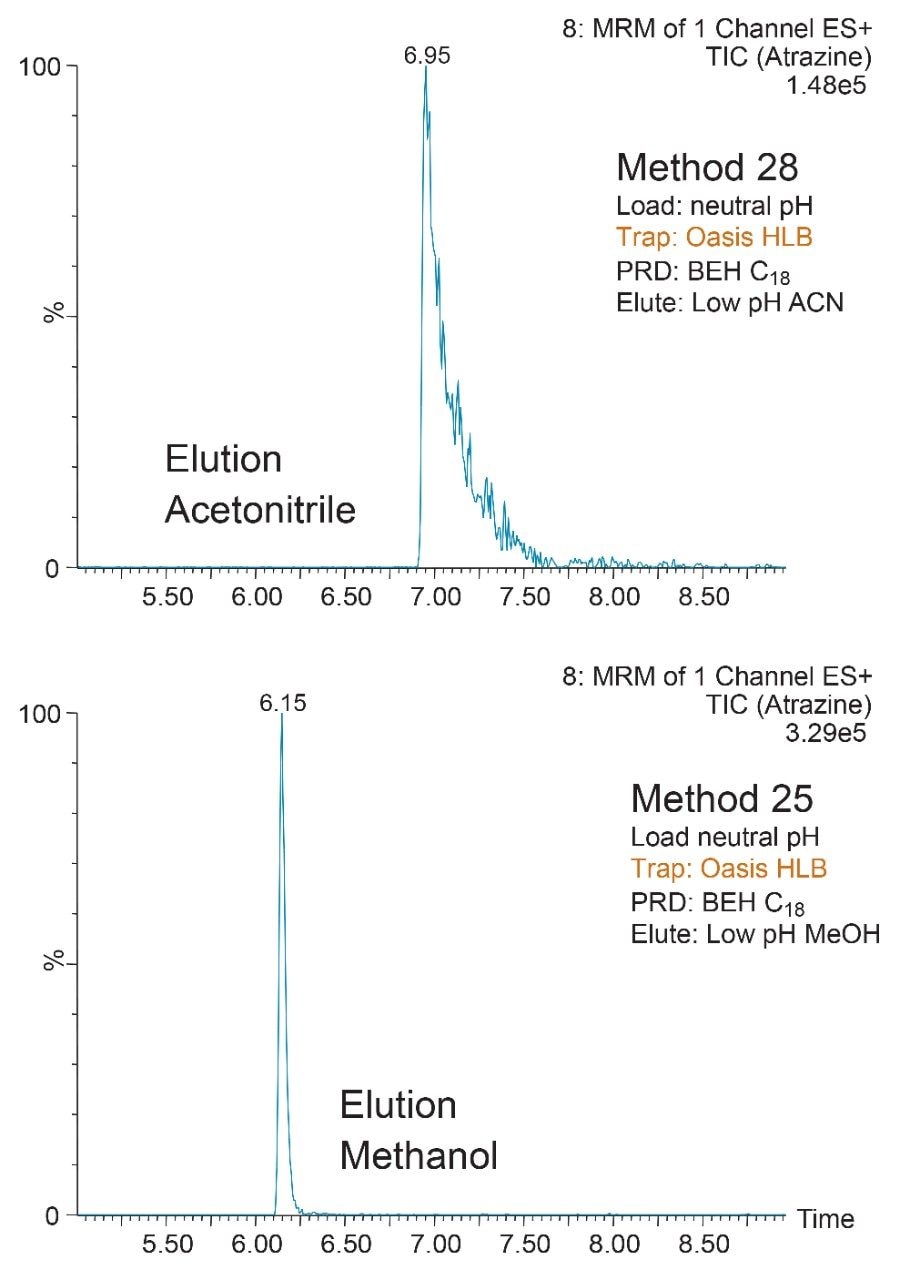
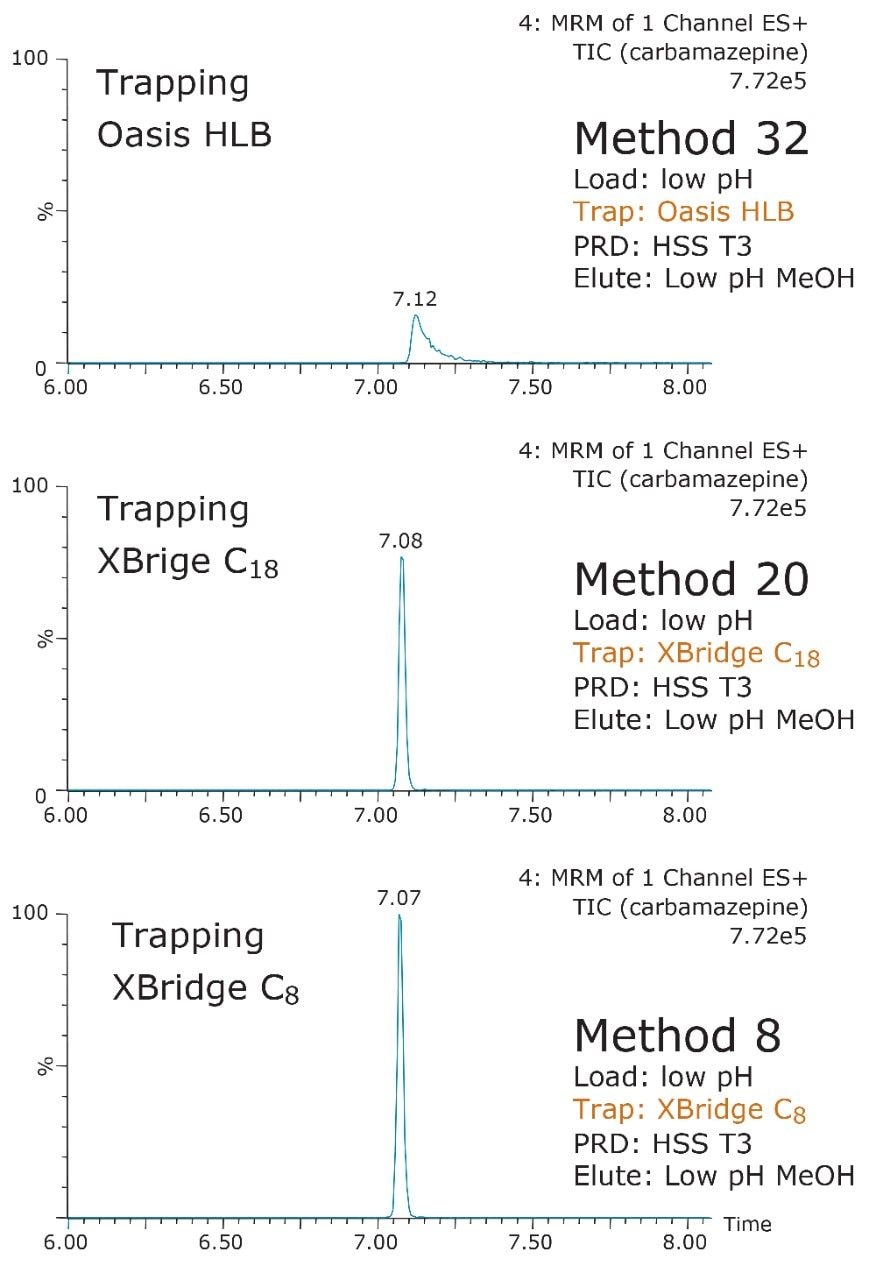
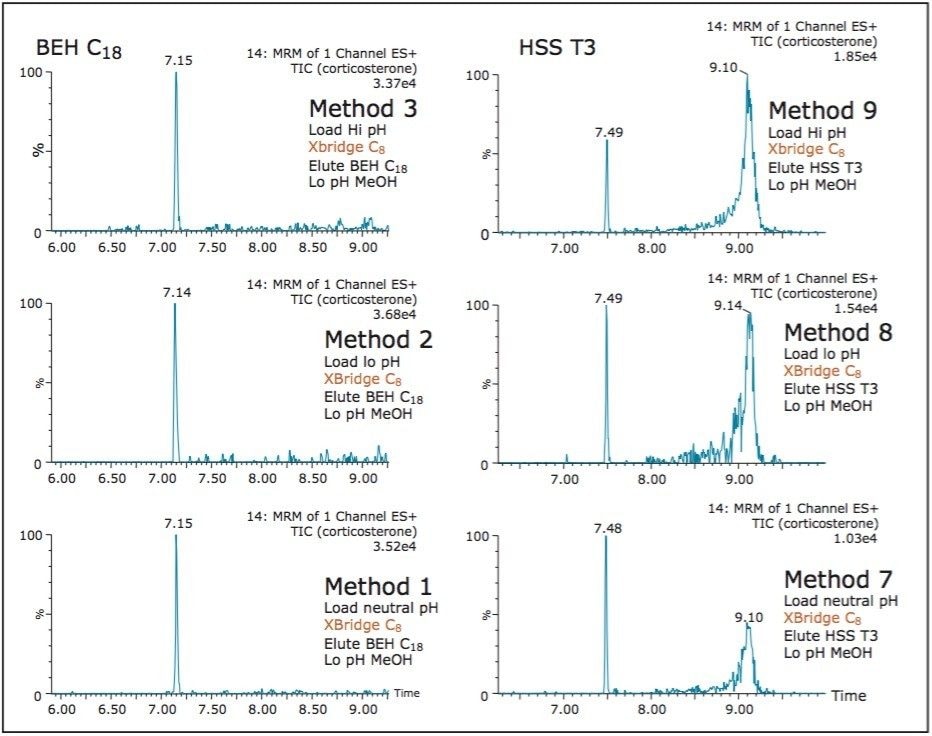
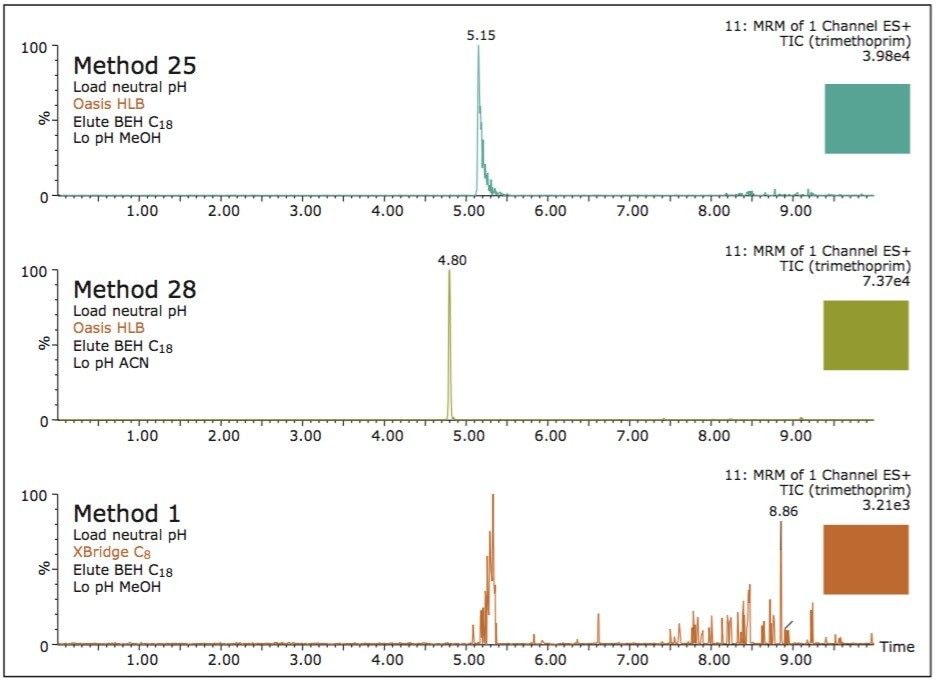
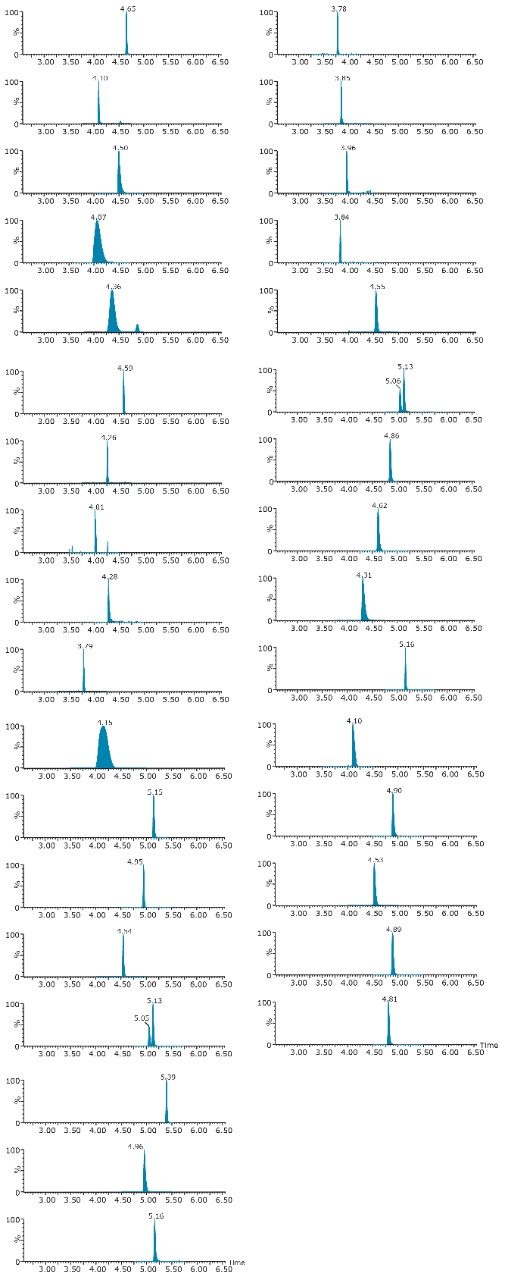
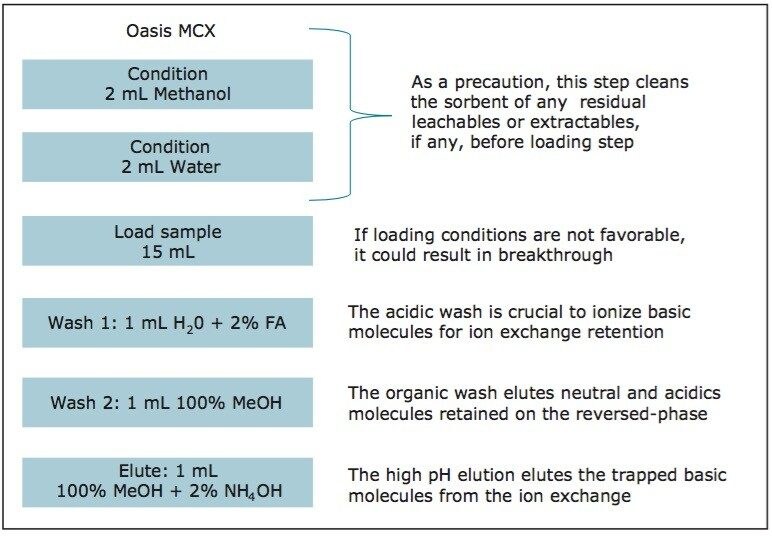
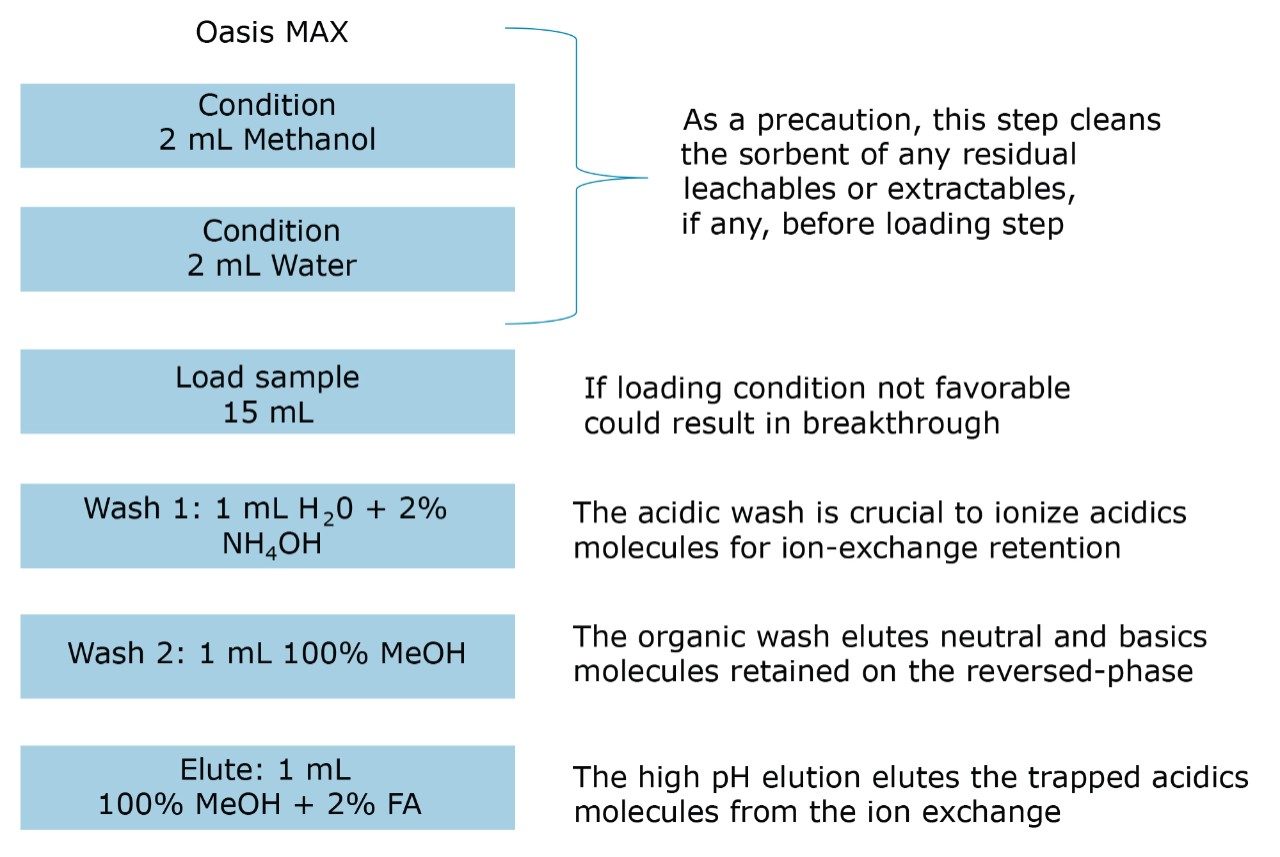
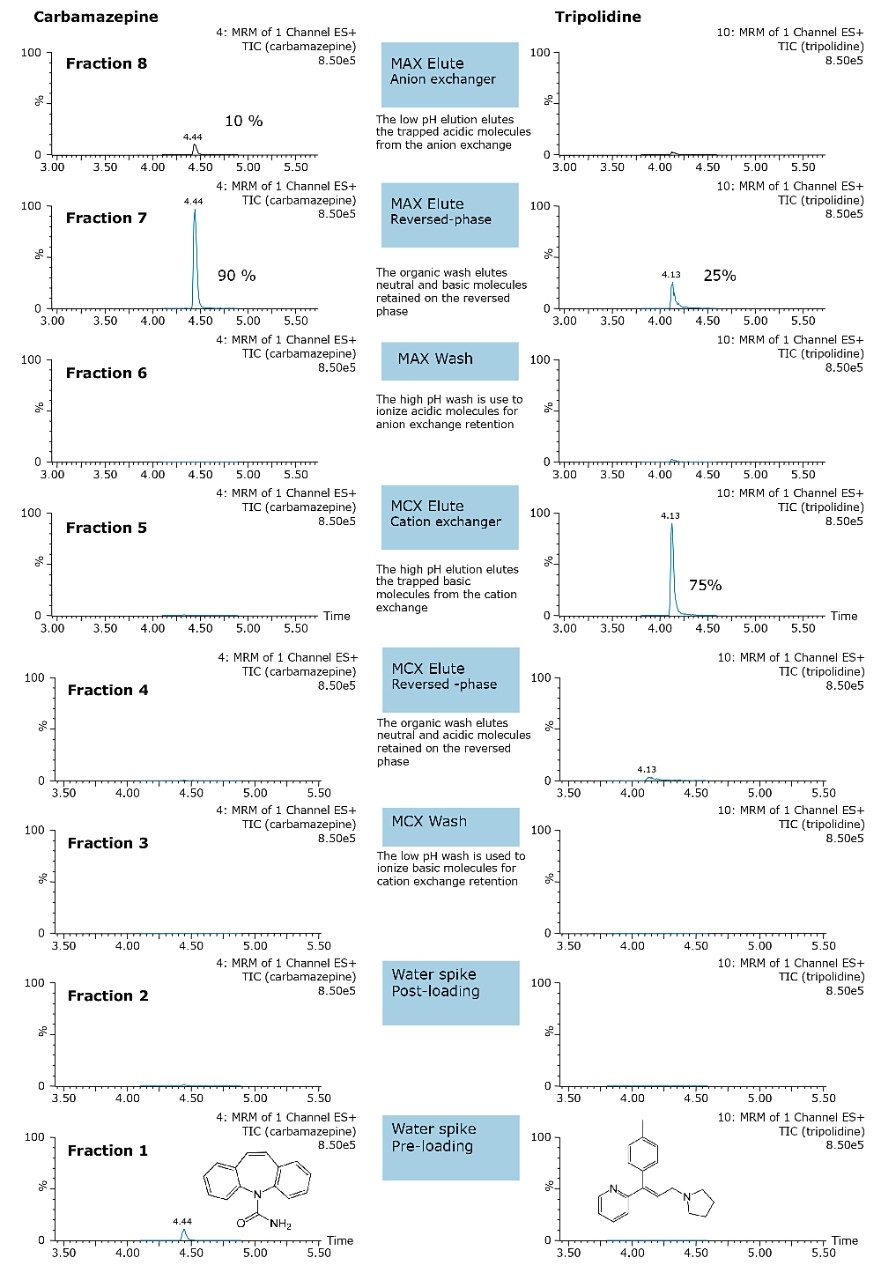
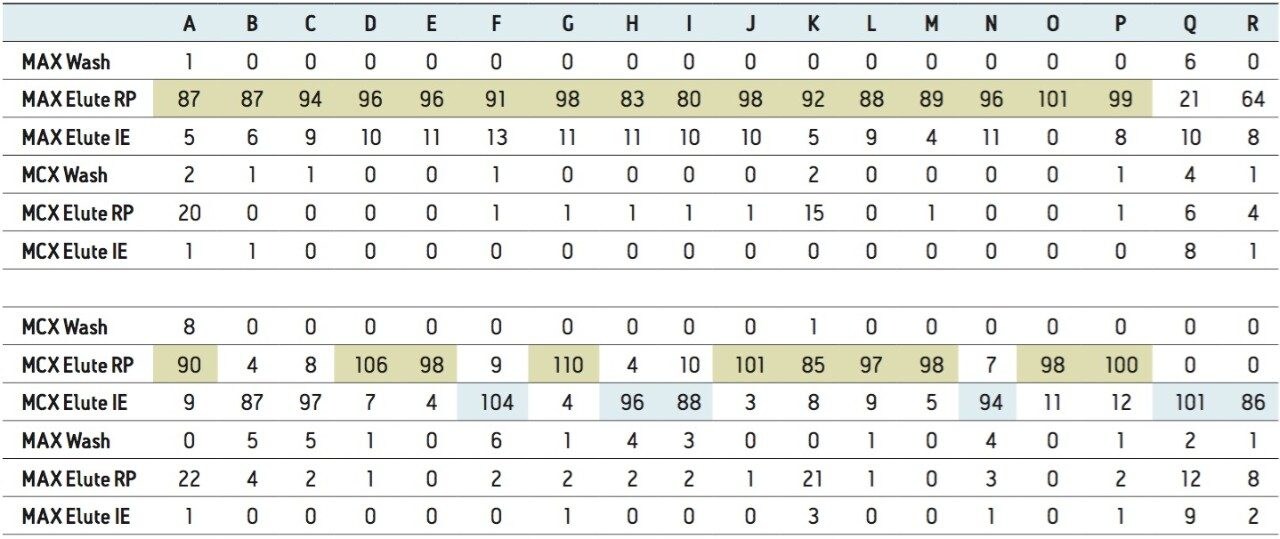
ACQUITY UPLC Systems with 2D-LC Technology offer the same analytical performances regarding recoveries, linearity, robustness, and lifetime, but at micro-extraction level. Figure 2 shows a micro extraction protocol using a 15-mL sample volume. The smaller sample volume allows faster loading time, on average less than 10 minutes. The final elution volume was optimized at 1 mL. The enrichment ratio for a micro extraction protocol is 15:1. With the option of a wider range of injection volume and extract composition, the evaporation and reconstitution step were eliminated. With 2D at-column dilution, aqueous and organic extracts can be loaded and captured on a trap column with high efficiencies. The injection volume for this configuration is not a limitation, which gives the option to inject as much as needed to reach target detection limits. For example, if the entire final sample (1 mL) is used for the analysis, it will give an additional 100:1 enrichment factor. Therefore, the overall enrichment from hardware and extraction protocol is now calculated at 1500:1, which is higher than those seen with a macro extraction protocol. Furthermore, the entire extraction protocol (loading, washing, and elution) was completed in less than 15 minutes for a high-grade, tap, and surface water sample.