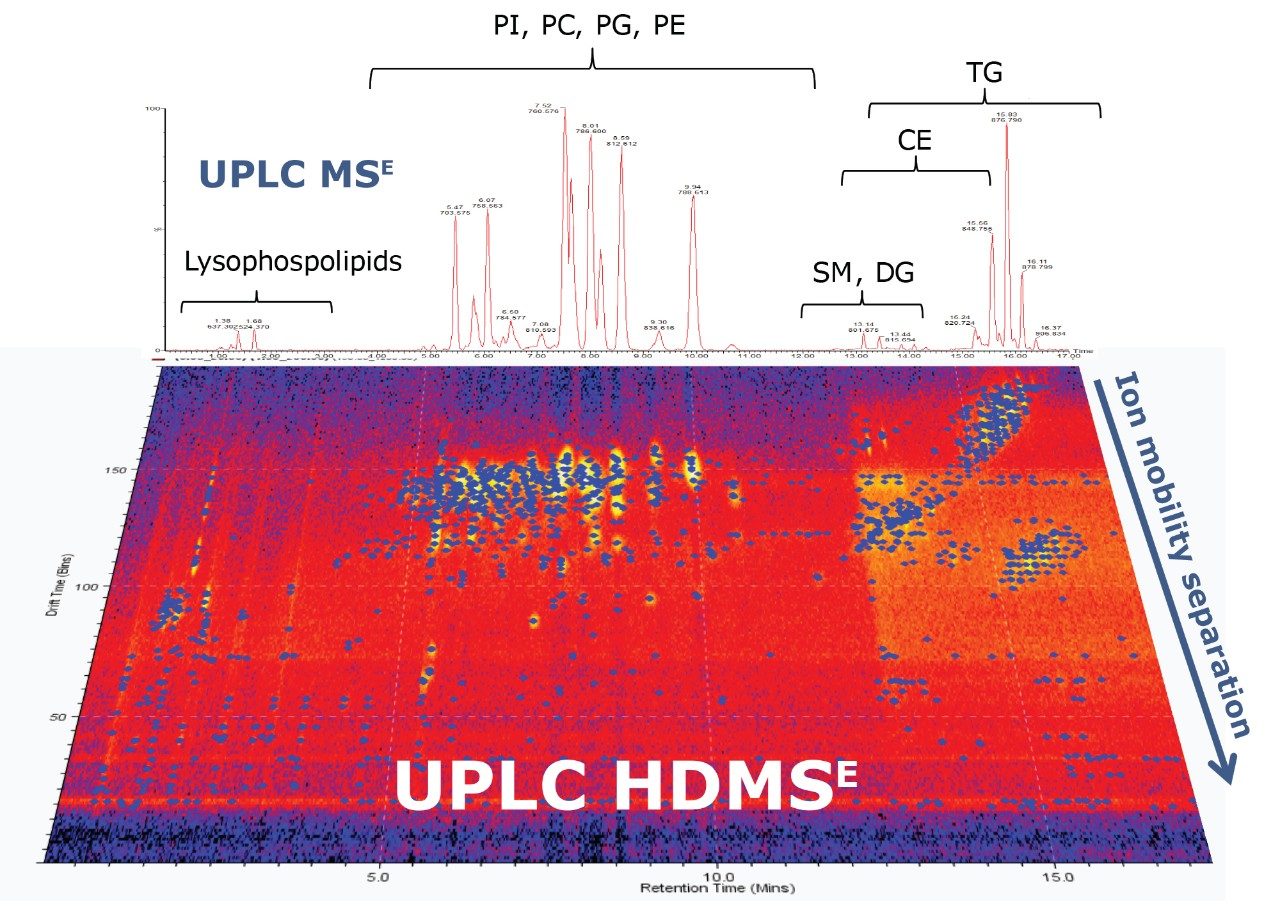
After chromatographic separation, lipids were ionized and entered in the mass spectrometer, where they passed through the Ion Mobility Separation (IMS) cell. A T-Wave mobility separator uses a repeating train of DC pulses to propel ions through the gas-filled cell in a mobility dependent manner. Lipids migrate with characteristic mobility times (drift times) according to their size and shape. For example, differences in the acyl chain length or number of double bonds affect the shape and size of lipid molecules, resulting in characteristic drift times.
Therefore, IMS provides an additional degree of separation besides chromatography, improving peak capacity over conventional UPLC, as shown in Figure 1. This leads to a better separation of lipid species and increased selectivity.
Finally, the lipid ions exiting the IMS cell were fragmented in the transfer T-Wave cell in MSE mode, which utilizes parallel low and elevated collision energy to acquire both precursor and product information for virtually every detectable ion in a single analytical run.4 The transfer T-Wave delivers the mobility separated ions to the time-of-flight mass analyzer, which records ion arrival times (or drift times).
The combination of IMS and MSE – known as HDMSE – provides increased specificity and hence confidence for lipid identification in complex biological mixtures, reducing false-positive identifications. HDMSE has several advantages over previous methods traditionally used to obtain the product ion data, such as multiple reaction monitoring or data dependent acquisition. Indeed, no prior knowledge of the lipid ions of interest is required and there is no loss of data due to poor duty cycle. Therefore, HDMSE acquisition is ideal for the rapid analysis of unknown lipid mixtures in biological samples.
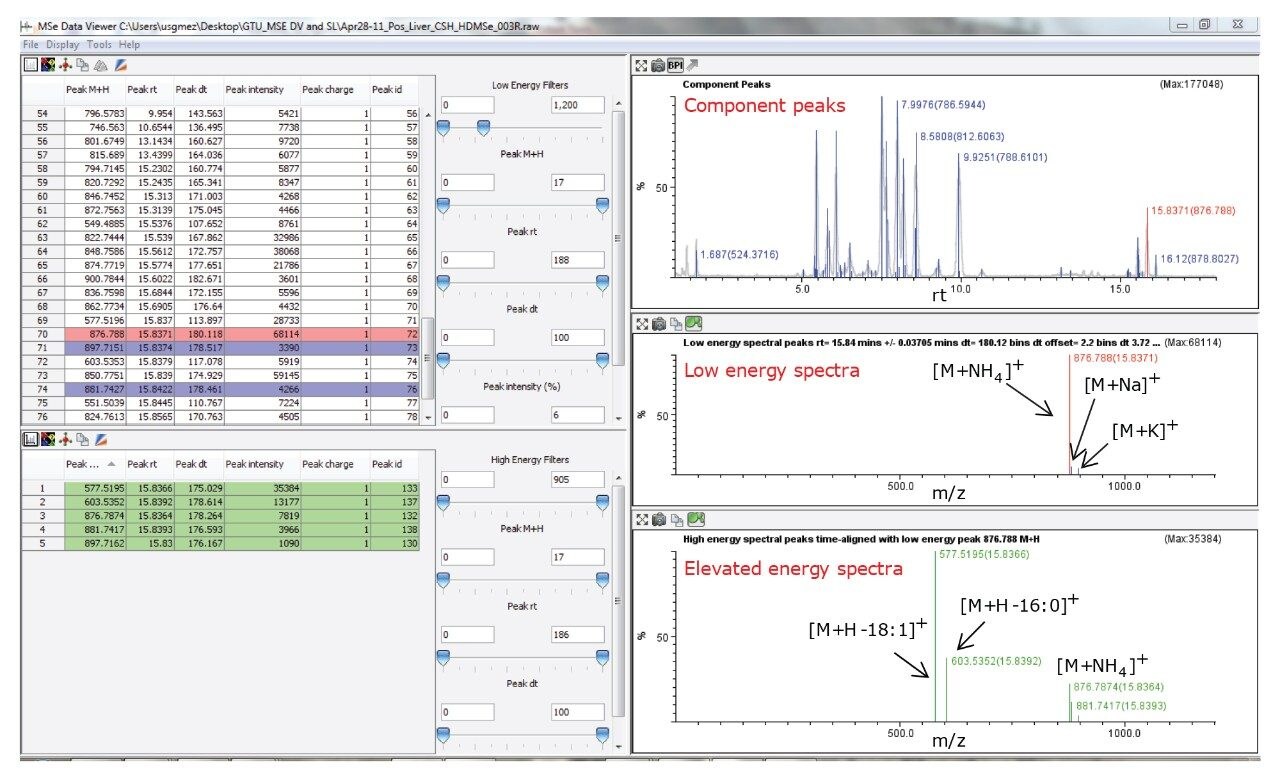
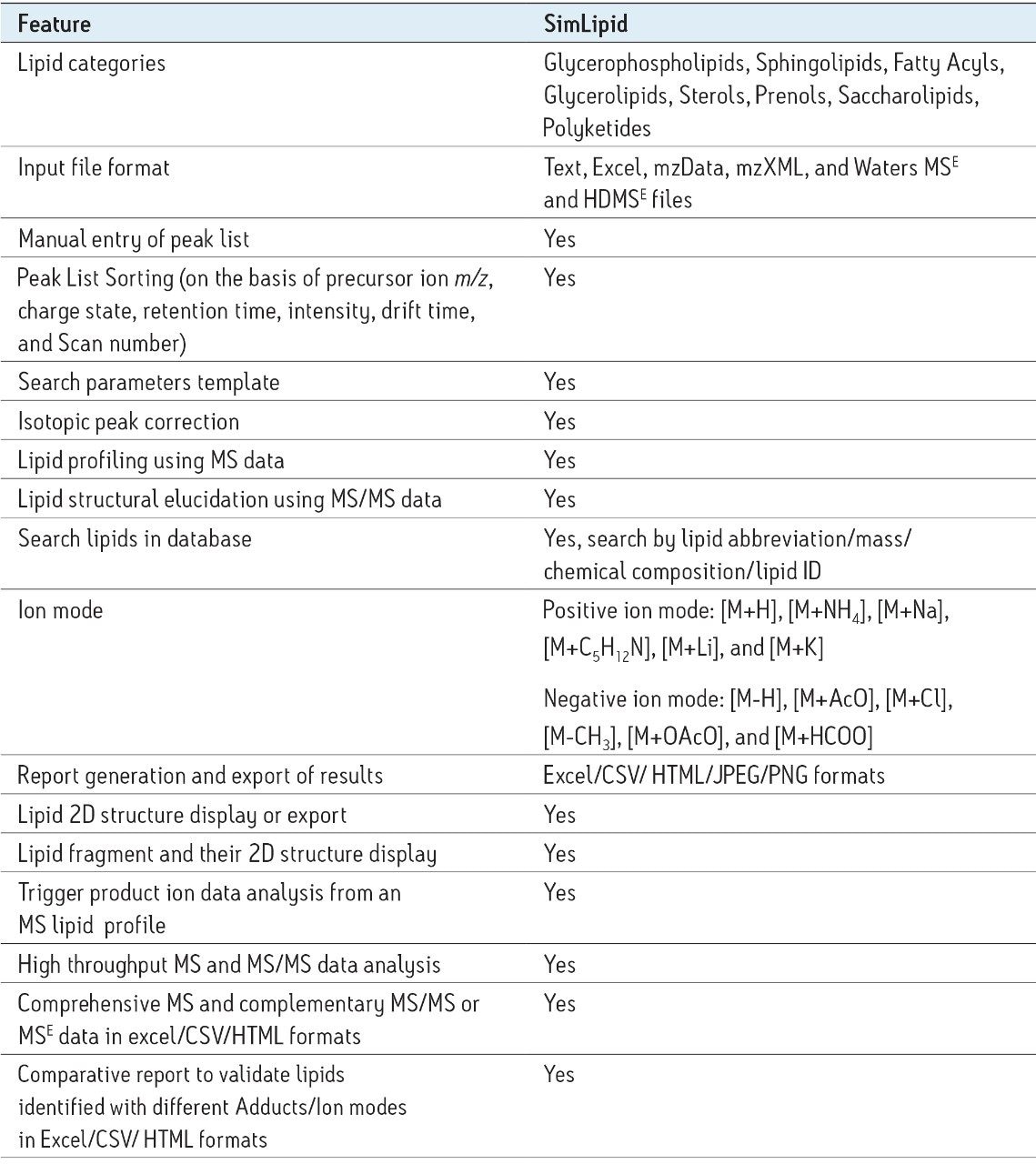
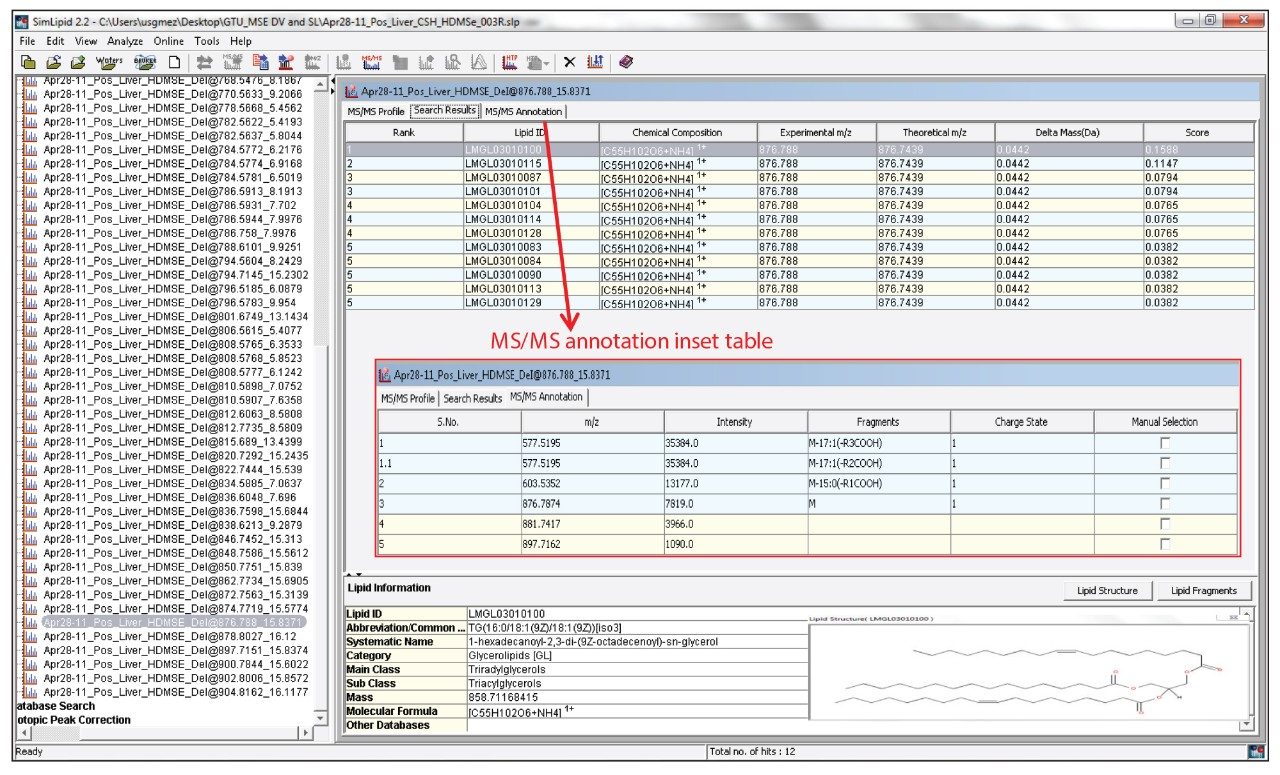
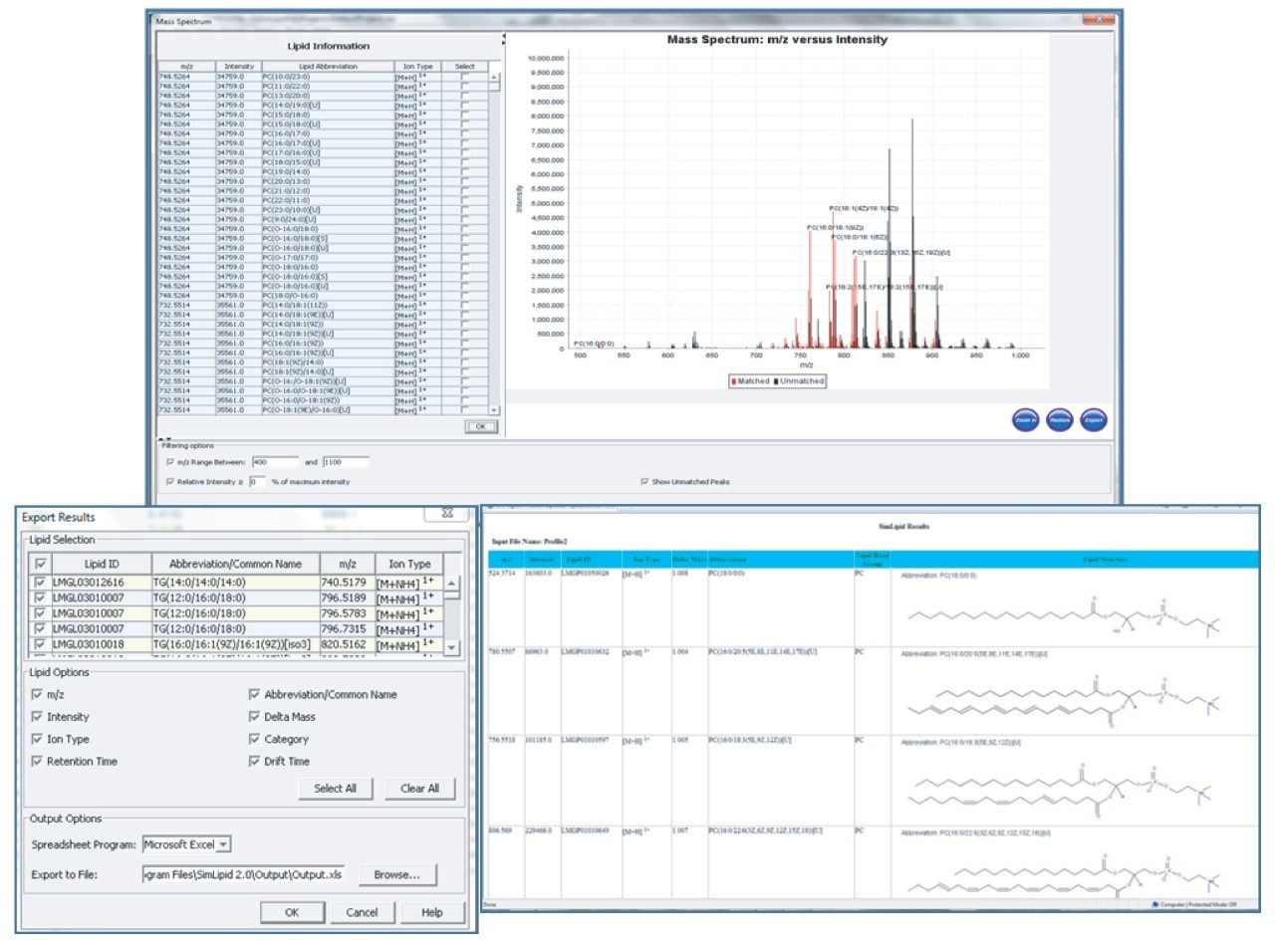
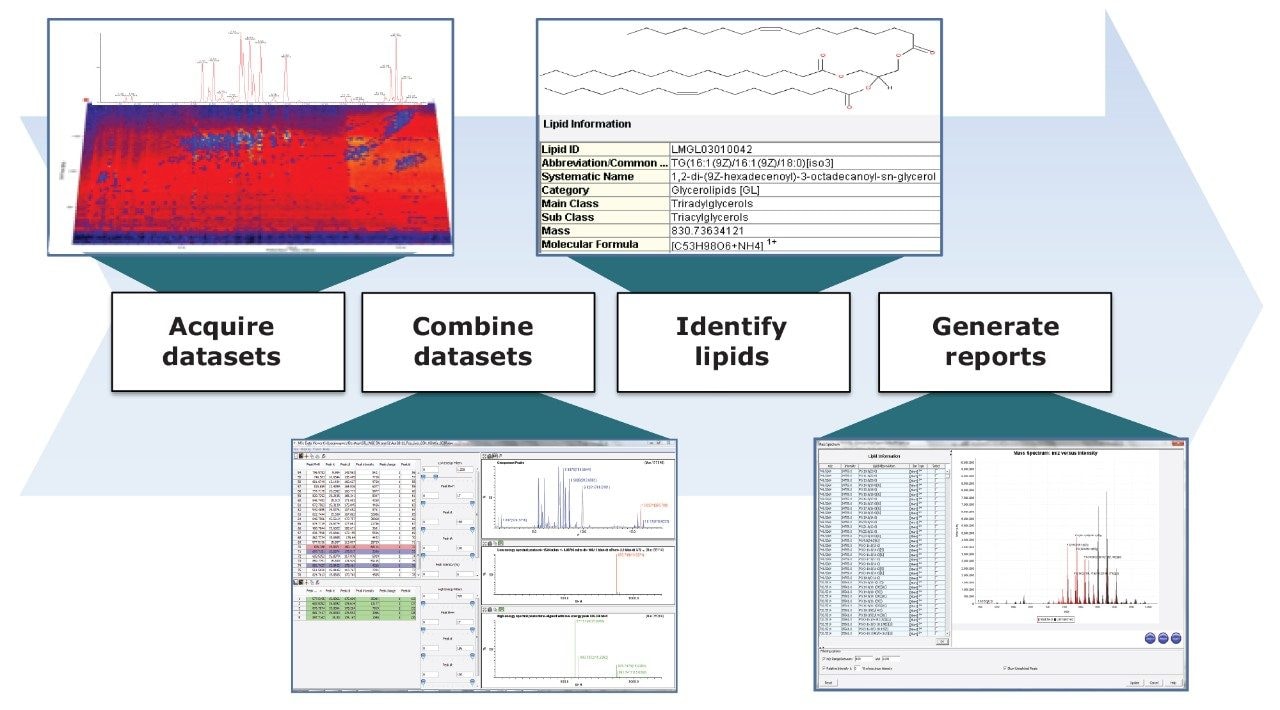
The data generated by UPLC-HDMSE was extracted using Waters MSE Data Viewer, software developed for visualization, processing, and interpretation of multi-dimensional MS or HDMS data, as shown in Figure 2. MSE Data Viewer uses a Waters proprietary algorithm, Apex 4D, to assign a unique retention time, drift time, m/z, and intensity to each individual lipid ion in the mixture. Precursor and product spectra are then aligned according to retention and drift times and linked together. An example of this application is shown in Figure 2, in which a lipid extract from bovine liver was analyzed by UPLC-HDMSE and MSE Data Viewer was used to process the datasets, which were then used for lipid identification through SimLipid Software.