Data quality analysis
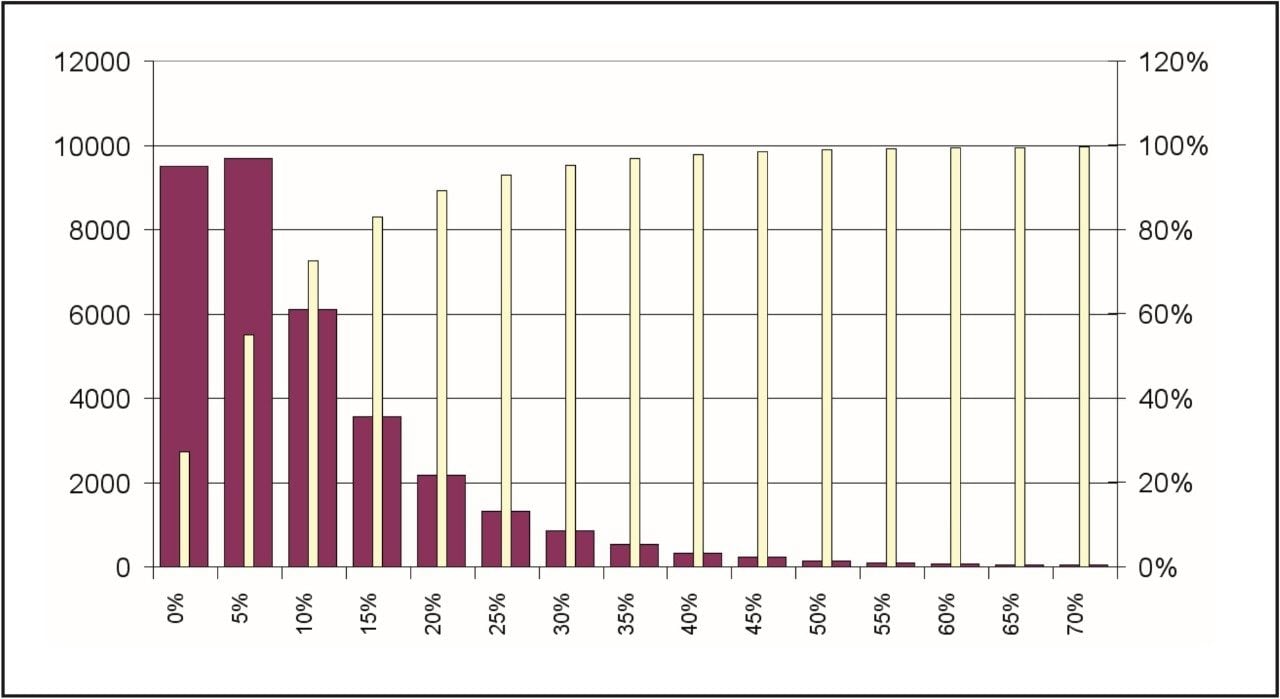
Figures 4 and 5 summarize the quality of the acquired data for the analysis of quantitative label-free proteomics experiments. Figure 4 shows the distribution of coefficient of variation of the accurate mass/retention time cluster intensities across all injection and conditions. The coefficient of variation of the majority (~ 70%) of the clusters – as indicated by the cumulative yellow bar – was found to be better than 15%, which indicates good consistency between injections and suitability for further quantitative analysis.
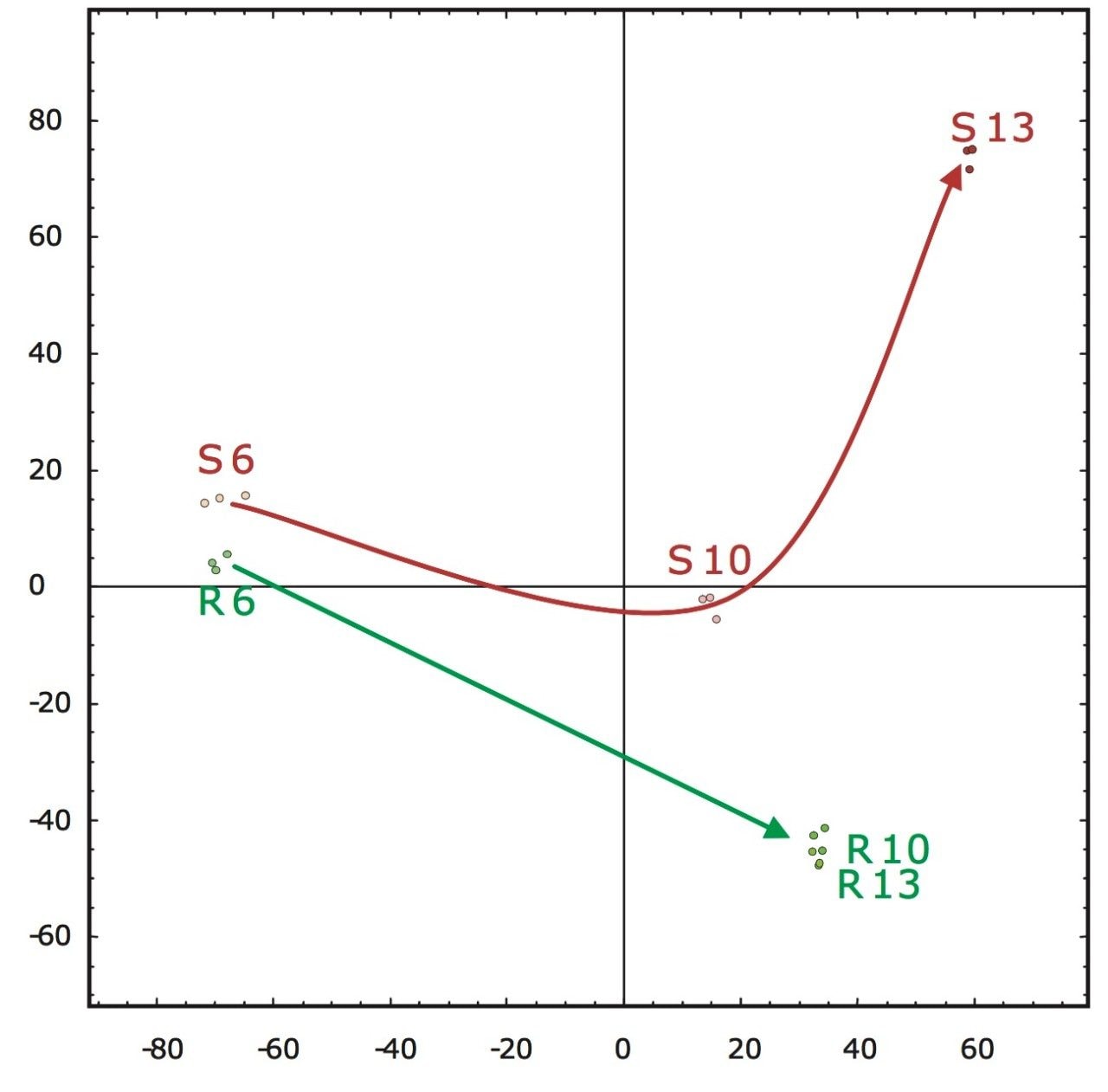
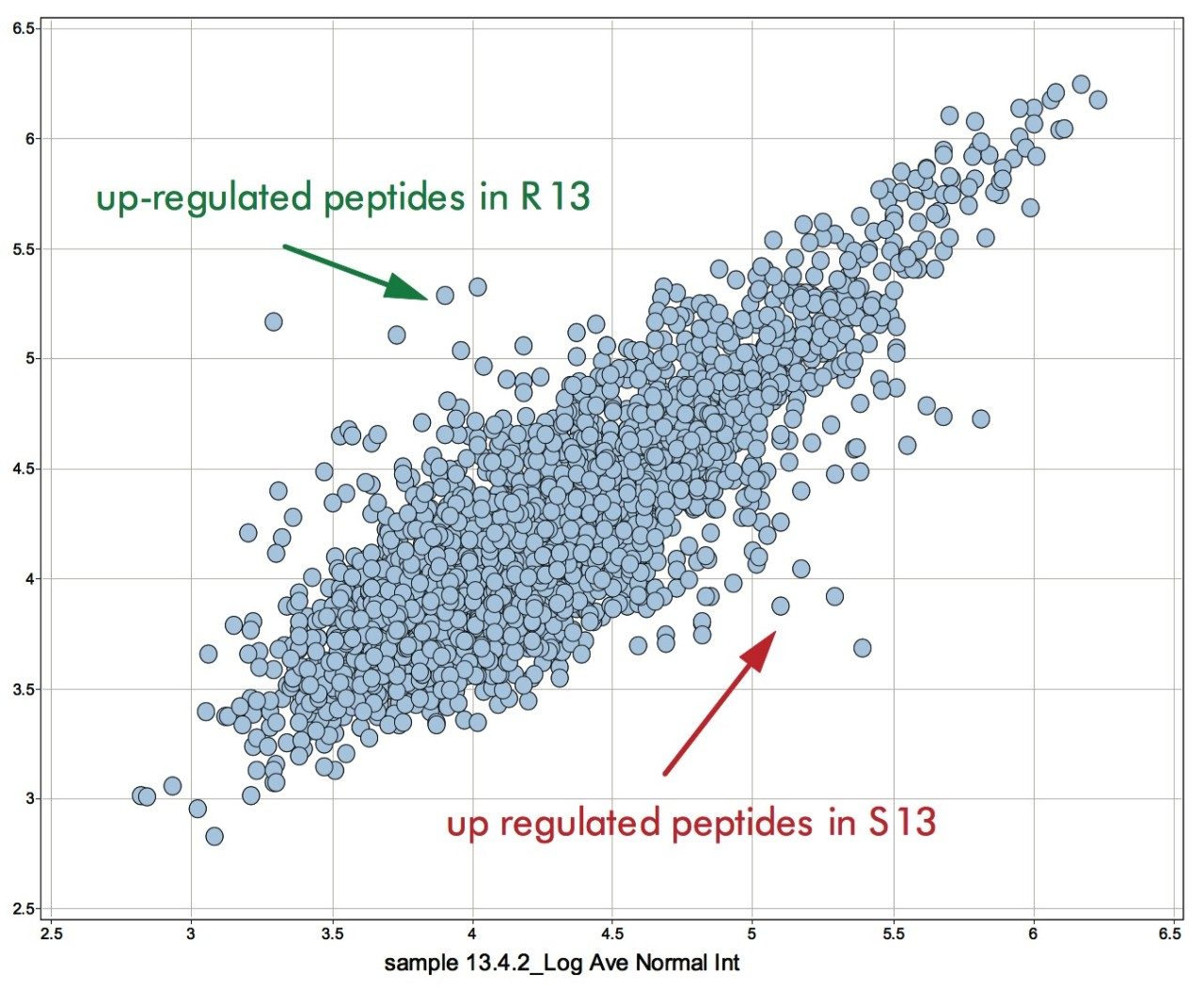
A second approach would be the analysis of the accurate mass/retention time pair clusters by means of principal component analysis (PCA) of which the final result is shown in Figure 5. The first two principal components can be related to incubation time (x-axis) and fungal growth (y-axis). The triplicate injections cluster extremely closely to one another, agreeing with the earlier observed reproducibility of the intensities of the peptides as shown in Figure 4.
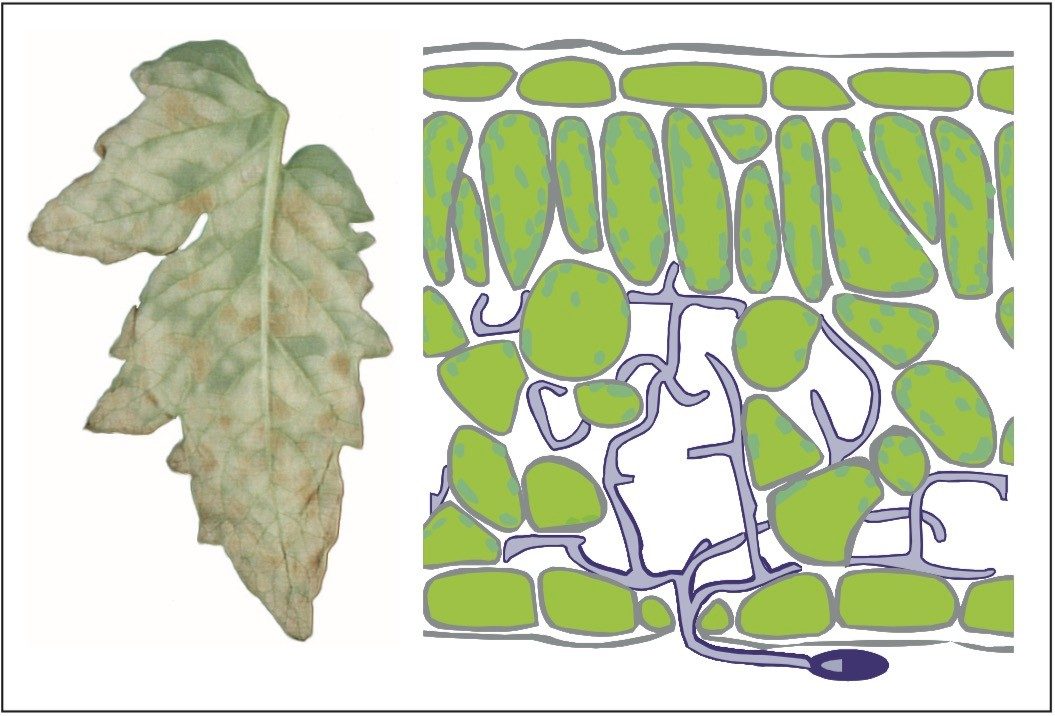
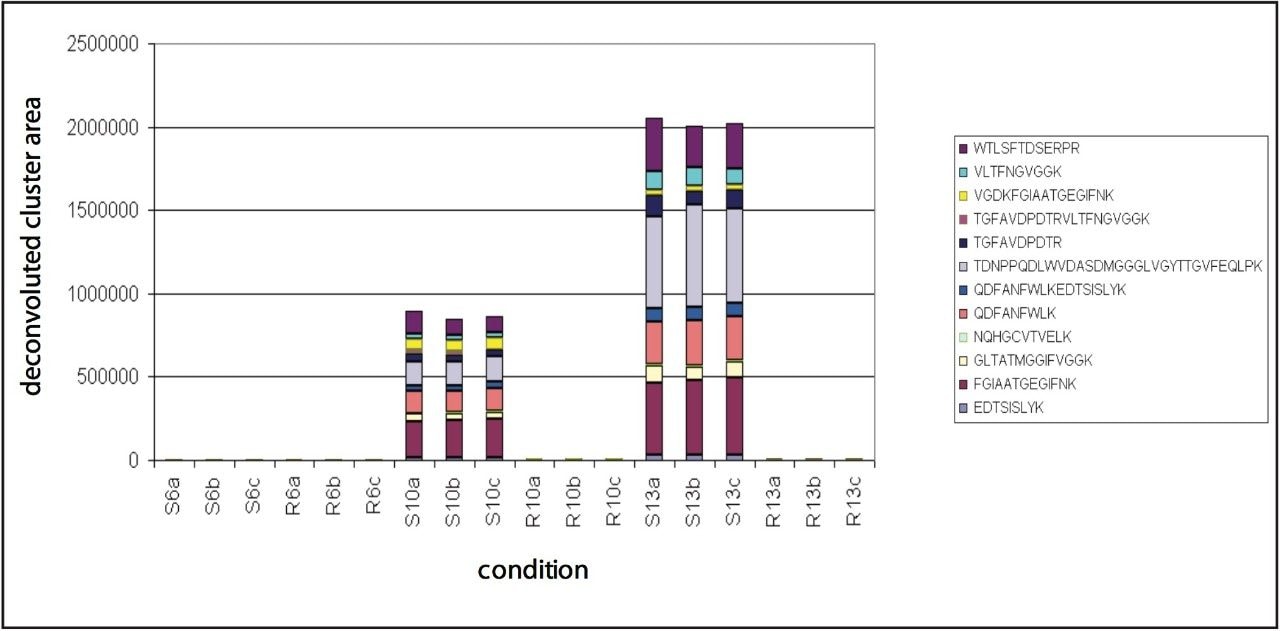
Figure 5 also illustrates that the samples from the resistant line are very similar in their peptide composition after 10 and 13 days of fungal inoculation. This is in agreement with biological findings as the fungus will not proliferate in the resistant tomato plant due to defense response after infection. This in contrast to the susceptible line, where protein changes continue after 10 days of inoculation as result of fungal growth. Many fungal secreted proteins are detected in the later time point samples S10 and S13. Again – for all samples – the replicate injections cluster closely together, indicating good data consistency.