Scaling of chromatographic methods is a routine necessity to utilize different instrumentation or column configurations for a successful separation method. A common example of this would be the fast development of a chromatographic method on the analytical scale with the goal of transferring the separation to the preparative scale. For liquid chromatography (LC) applications, this scaling methodology is well understood and the guidelines for transferring methods are straight forward. For chromatographic methods using CO2 as the principal component of the mobile phase, the scaling process is not as well understood. This is due to the high compressibility of the CO2 mobile phase which makes many of the scaling methodologies developed for LC invalid. Therefore, most of the current scaling strategies used for supercritical fluid chromatography (SFC) are based on empirical observations and often times require additional method manipulation on the transferred system.
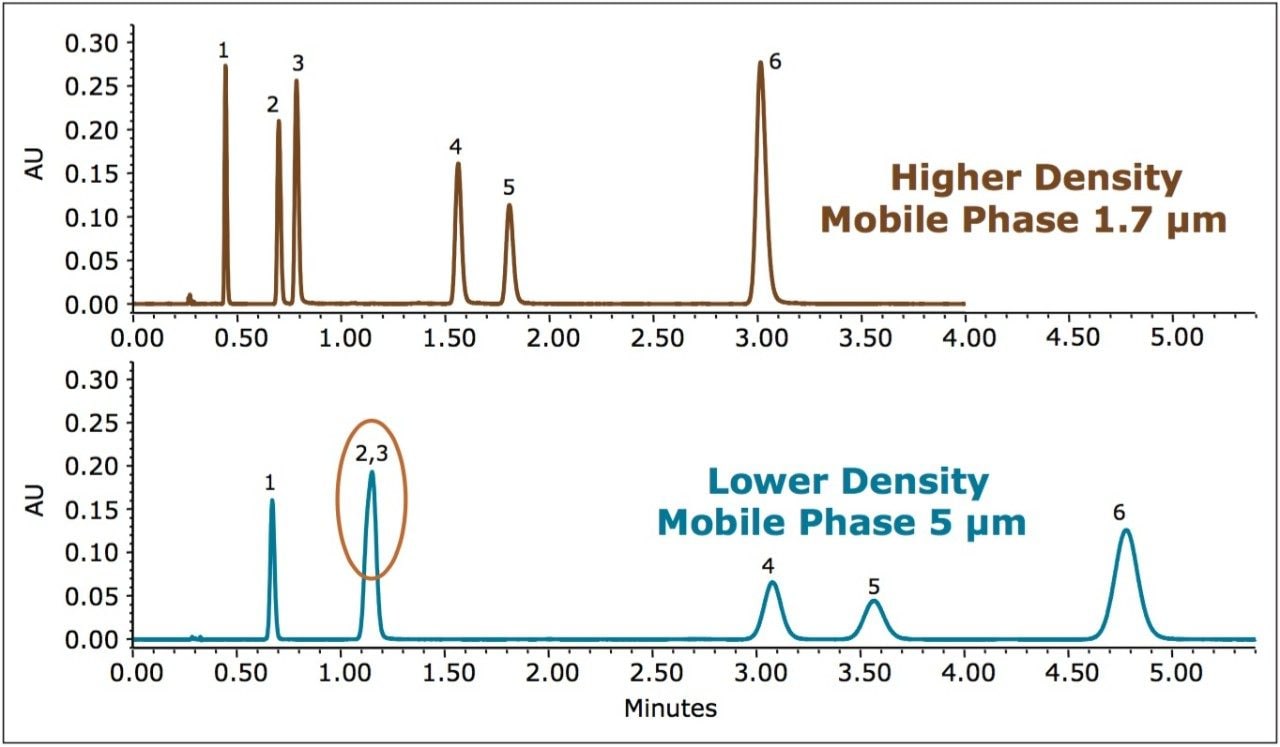
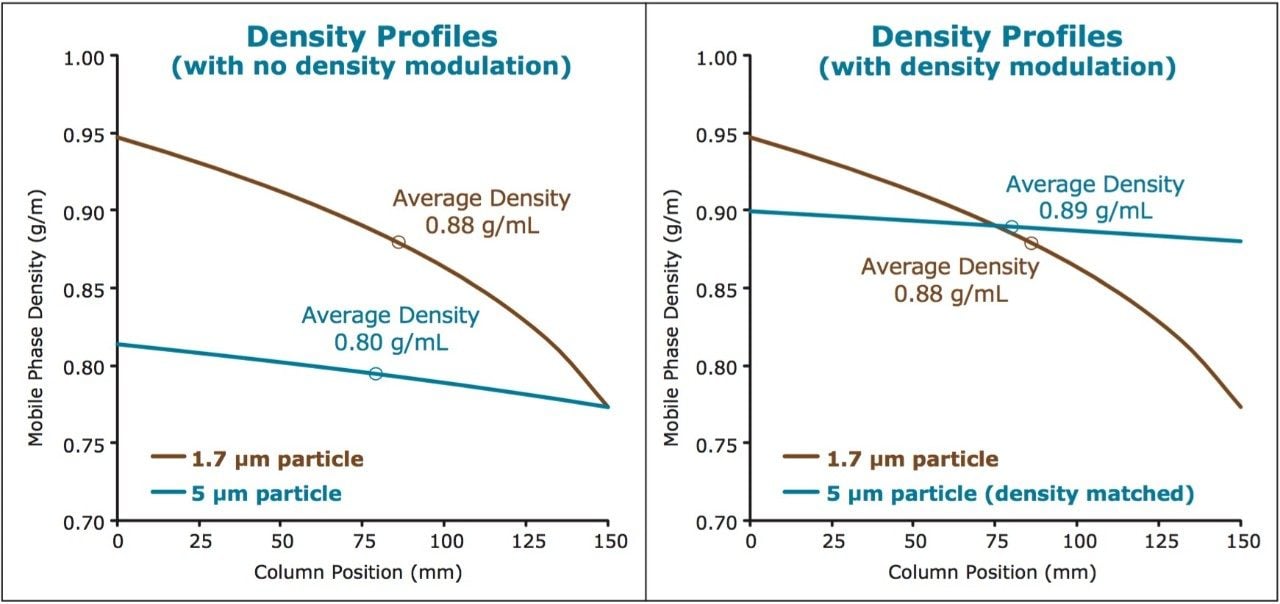
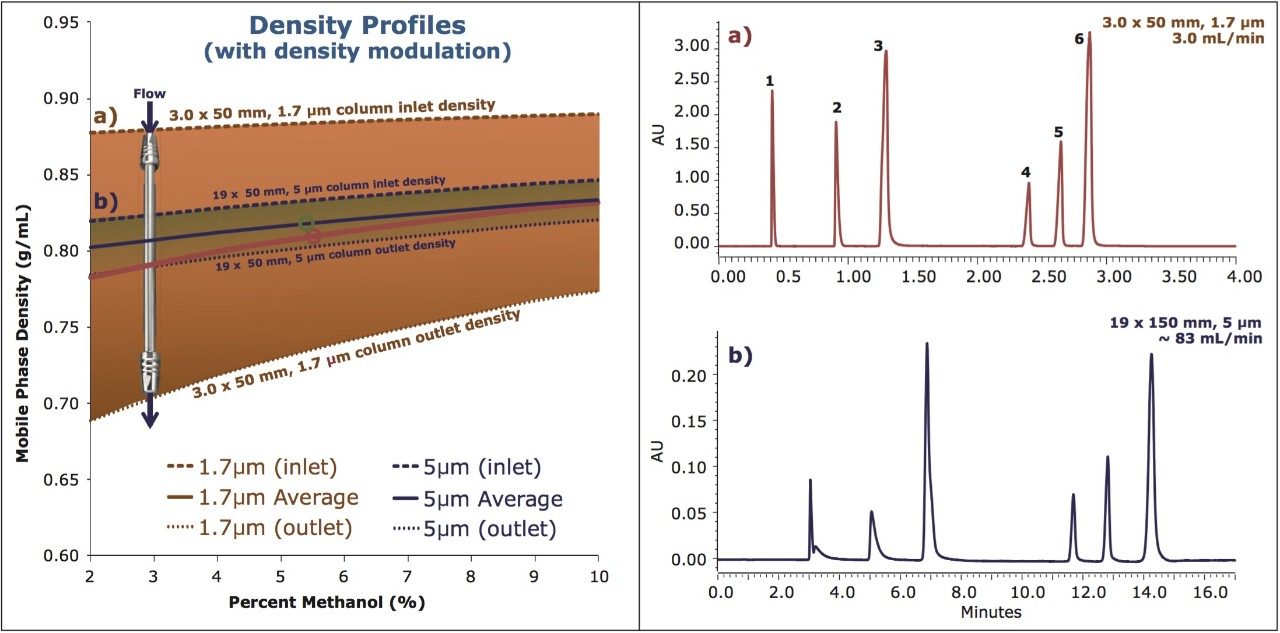
It is well known that for separations using CO2 as the principal mobile phase component, analyte retention factors are influenced largely by the mobile phase density and temperature. Because of the high compressibility of CO2 under standard operating conditions, the density can change significantly with changes in pressure (under isothermal conditions), with retention factors increasing with decreasing mobile phase density (pressure). In addition, the selectivity and resolution of the analytes may be impacted as they respond differently to the same changes in mobile phase density. This can present a challenge when attempting to transfer a method between different column configurations that involve changes in column length or stationary phase particle size, which in turn alters the pressure (density) profile along the column. This is best exemplified when analytical scale separations, developed using UltraPerformance Convergence Chromatography (UPC2) on sub-2-μm stationary phases, are scaled up for preparative SFC conditions using 5 μm particle size stationary phases. The difference in the density profiles across the column, between the analytical and the preparative system, may lead to very different chromatography unless the scale-up procedure is guided by a systematic approach.
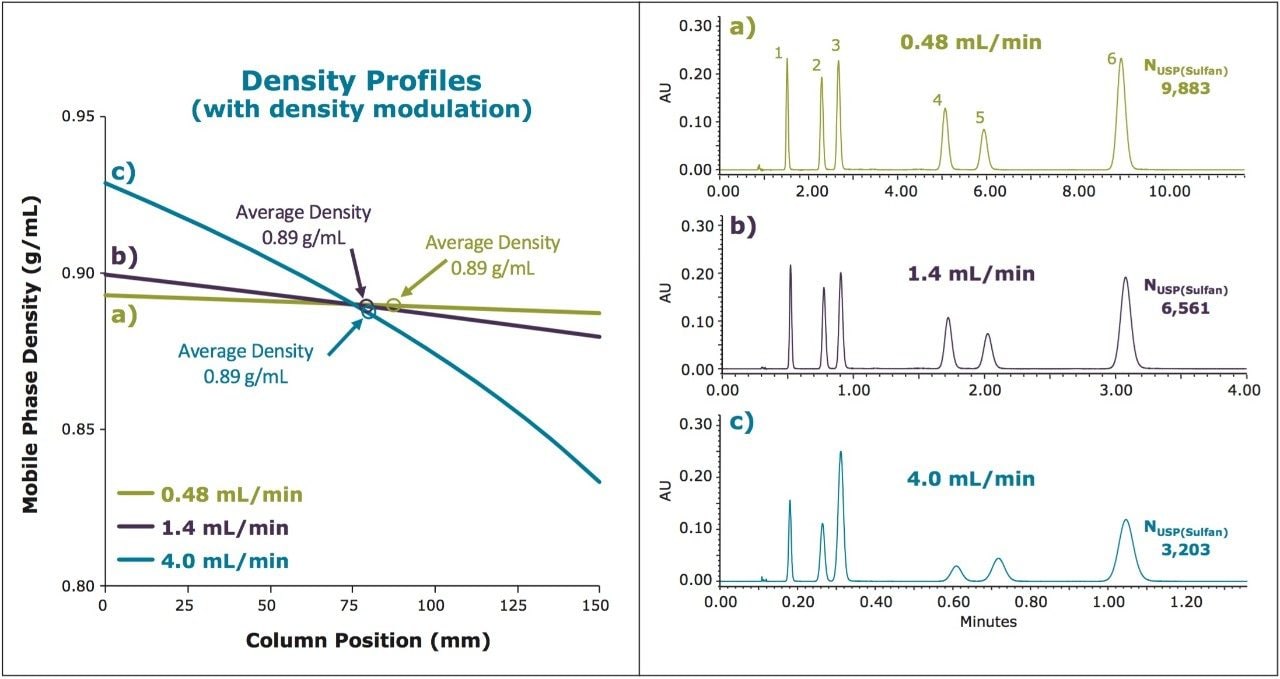
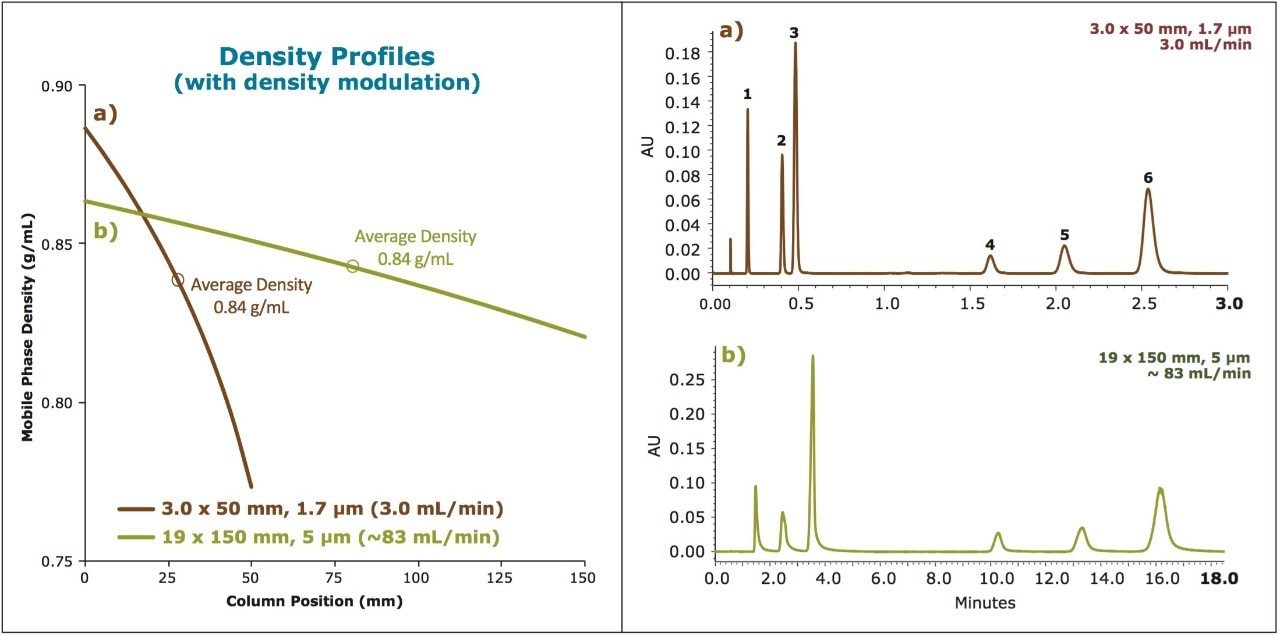
Here we present a strategy for scaling SFC separations between various system, condition, and column configurations by employing density modulation to maintain similar average density profiles between separations. The ability to scale methods efficiently enables the rapid screening of methods on the faster analytical scale (using UPC2), with the direct transfer of the final method to preparative chromatography while maintaining chromatographic integrity between separations. The net result is a scalable, predictable separation with significant savings in time and mobile phase costs (raw materials and disposal of waste).