Rapid and Sensitive Detection of dsRNA Contaminants in mRNA Using the GTxResolve™ 250Å Slalom Chromatography Column
Jamuna Vaishnav, Matthew Lauber, Balasubrahmanyam Addepalli
Waters Corporation, United States
Published on August 29, 2025
Abstract
Discovery and development of messenger RNA (mRNA) drug substances has revolutionized both preventive (vaccines) and curative (cancer immunotherapy, and defective protein replacement) therapies. A critical step in mRNA synthesis is in vitro-transcription (IVT), a process that makes single-stranded mRNA from a DNA template. However, this process often produces immunostimulatory double-stranded RNA (dsRNA) contaminants that compromise therapeutic safety and efficacy. This application note introduces a highly sensitive and rapid two-step analytical method for dsRNA detection. In the first step, the single stranded mRNA is treated with RapiZyme™ Cusativin to decrease drug substance ssRNA interference. The second step entails advanced separation capabilities of the GTxResolve 250Å Slalom Chromatography Column to resolve dsRNA from degradation products of ssRNA. Sensitive detection of dsRNA is also facilitated by the hydrophilic high performance surfaces and advanced packing material of chromatography columns. Compared to traditional agarose or capillary gel electrophoresis, slalom chromatography requires ~40X less dsRNA sample (10 ng vs ~400 ng for agarose gel) and takes <2h for detection compared to other traditional methods. Such a robust identification and quantification approach for dsRNA offers a faster and more efficient alternative analytical method to help fine tune mRNA drug substance formulations so that the safety, quality, and regulatory compliance of mRNA-based therapeutics is ensured.
Benefits
- Direct detection of low levels dsRNA in single stranded mRNA sample is achieved
- Combined enzymatic and chromatographic workflow is a promising approach for sensitive, specific dsRNA analysis
- RapiZyme Cusativin selectively digests ssRNA not dsRNA
- The GTxResolve 250Å Slalom Chromatography Column enables high-resolution separation of dsRNA species from degradation products of ssRNA
- Excellent analyte recovery with no nonspecific adsorption
Introduction
mRNA therapeutics are transforming the landscape of modern medicine, offering a versatile platform for development of vaccines, protein replacement, and cancer immunotherapies. With the recent success of mRNA-based COVID-19 vaccines, interest in this technology has surged, fueling large-scale development and manufacturing efforts across the globe. At the heart of this technology lies IVT, an enzymatic process that enables the production of high-yield, functional single-stranded mRNA (hereafter referred to as ssRNA) molecules from a DNA template.1 While IVT is efficient and scalable, it is not without challenges. A critical concern facing developers is the presence of dsRNA contaminants—unintended byproducts that can significantly compromise product safety, immunogenicity, and efficacy. dsRNA contaminants arise through erroneous 3’ extension where rebinding of product RNA to T7 RNA polymerase is followed by self-priming to generate duplex RNA. This process occurs most commonly under high yield reaction conditions by successfully competing with the promoter driven RNA synthesis.2 Even if the contaminant dsRNA was present at low levels, their impact is disproportionately high as trace amounts of dsRNA are known to activate innate immune receptors such as toll-like receptor 3 (TLR3), RIG-I, and MDA5, potentially triggering unwanted immune responses.3
Several analytical methods including agarose gel electrophoresis, enzyme-linked immunosorbent assay (ELISA), and dot-blot assays are used to detect dsRNA contamination in IVT reactions. Agarose gel electrophoresis offers a simple approach but suffers from low sensitivity. ELISA, leveraging dsRNA-specific antibodies, provides high sensitivity and reliability; however, it involves multiple steps and requires several hours to complete.4 Similarly, dot-blot5 and immune-northern techniques6— based on monoclonal antibody recognition— are time-intensive and prone to false positives due to nonspecific adsorption on membranes. Given these limitations, there is a pressing need for a rapid, user-friendly, quantitative, and highly sensitive technique for the accurate detection of dsRNA.
Slalom chromatography emerges as a promising alternative for the separation of large nucleic acids due to its unique reliance on shear forces generated by pressure-driven flow rate rather than electrophoretic forces.7 This technique offers high-throughput capabilities, enabled by its inherently fast flow rates and efficient separation dynamics. Relying on shape, length, and nucleic acid type dependent stretching and relaxation kinetics, slalom chromatography can be used for a multitude of nucleic acid analytical purposes. Compared to traditional methods like gel electrophoresis, slalom chromatography provides superior resolution and detection efficiency, making it a powerful tool for nucleic acid analysis. The GTxResolve 250 Å Slalom Chromatography Column engineered with inert hardware and advanced packing material was previously shown to analyze large nucleic acids8 and plasmid quality9 in under five minutes. This column is also capable of providing a tunable analytical method where key parameters such as flow rate, solvent viscosity, and column temperature can be adjusted to optimize performance.
This application note provides a preliminary investigation into analytical workflow for sensitive and selective dsRNA detection. The two step process involves enzymatic digestion of an mRNA sample with RapiZyme Cusativin10, an endonuclease that selectively degrades ssRNA while leaving dsRNA intact. This enzymatic discrimination allows researchers to enrich the sample for dsRNA with high specificity. This is followed by sensitive and reproducible dsRNA detection where an optimally packed and rigorously tested GTxResolve 250Å Slalom Chromatography Column separates the intact dsRNA from the degraded products of ssRNA. The combination of selective enzymatic digestion and high-performance chromatography enables confident identification and quantification of dsRNA, even at low abundances. Notably, the method requires >40-fold less dsRNA samples compared to conventional electrophoretic techniques offering a faster, high-throughput scalable solution in performing process development and quality control assessments for safe deployment of mRNA therapeutics.
Experimental
|
1X TAE buffer: |
40 mM Tris-acetate, 1 mM EDTA pH 8.3 made from 10X TAE (Thermo Fisher Technologies p/n: 15558042) |
|
Column storage: |
10% acetonitrile, 90% aqueous 25 mM Sodium Phosphate pH 7.0 + 100 mM Potassium Chloride |
|
Seal and weak wash: |
20% Methanol: 80% Water |
|
dsRNA: |
Model dsRNA was made by annealing sense and antisense single stranded RNAs from a 3,000 nucleotide stretch of cas9 gene. DNA templates for RNA synthesis were made by polymerase chain reaction (PCR) from a 3 kb portion of cas9 gene using Cas9 plasmid (Millipore Sigma p/n: CAS9P) as source. PCR was done by using Q5 High-Fidelity 2X Master Mix (NEB Cat# M0492S) as per manufacturer’s instructions. The T7 promoter sequence was incorporated into forward PCR primers of either sense strand (Template 1) or antisense strand (Template 2) to drive the transcription. Run-off IVT was done for both DNA templates separately by using T7 polymerase (YEASEN, Cat# 10628ES10), 7.5 mM NTP (NEB Cat# N0466S), murine RNase inhibitor (NEB Cat# M0314L) and inorganic pyrophosphatase (NEB Cat# M2403L). Equal concentrations (1:1) of the synthesized sense and antisense RNA products (100 ng/µL each) were combined with 2 µL of 10X annealing buffer (1X) in a 20 µL reaction. The synthesized RNA was purified by Monarch® RNA Cleanup Kit (NEB p/n: T2040L) following treatment with TURBO DNase™ I (Invitrogen p/n: 4022G). To facilitate annealing, the RNA mixture was heated to 95 °C for 2 minutes to denature any secondary structures followed by shifting to 70 °C for 10 minutes and gradual cooling to room temperature over 30 minutes, allowing the complementary RNA strands to hybridize and form dsRNA. The purified dsRNA was stored in aliquots at –20 °C to ensure stability and prevent degradation. |
|
RNA digestion with RapiZyme Cusativin (Waters p/n: 186011192): |
The 20 µL digestion mix consisted of 100 ng of ssRNA or dsRNA, 200U cusativin and 100 mM ammonium acetate buffer (pH 9.0). The enzyme levels need to be scaled up depending on the amount of ssRNA in the sample. The mixture was gently vortexed, briefly centrifuged, and incubated at 30 °C for 1 hour. Enzyme activity was terminated by heating the samples at 75 °C for 15 minutes. |
|
Agarose gel electrophoresis: |
About 0.6 g of agarose (Sigma Aldrich p/n: A0539-500G) was dissolved in 100 mL of 1X TAE buffer by boiling in a microwave. After a brief cooling (~1–2 minutes), 10 µL of SYBR™ Gold Nucleic Acid gel stain (Invitrogen p/n: S11494) was added, thoroughly mixed and poured into a gel tray with a comb for solidification at room temperature. 1X TAE buffer was added on top of the solidified gel and the comb was gently pulled to make wells for sample loading. The gel setup was placed in a horizontal Bio-Rad electrophoresis system and required sample amounts were loaded into individual wells. Electrophoresis was conducted at 50V for 2 hours and 10 minutes after connecting it to the PowerPac™ HV High-Voltage Power Supply. The gel was subsequently imaged using the Gel Doc™ EZ Gel Documentation System (Bio-Rad) to visualize the bands. |
|
LC system: |
Waters ACQUITY™ Premier System (or equivalent that support high column pressure) consisting of QSM H-Class (Quaternary Solvent Manager with 50 µL mixer), TUV Detector - 1000 psi MAX Pressure 70 bars Max Pressure (p/n: 205015016) Sample Manager FTN (Ver. 2023 03 02), 15 µL Needle size (p/n: 700012820) Assy, Needle, FTN-15 µL, HPS, TXT w/Gd&st), 100 µL Sample syringe size, Injection Valve Flow Through (p/n: 700011791) External Pre-Heater, CH-30A, with Active Preheater (APH) MP35N, 0.004” ID, 18.5’’ (p/n: 430005558) and post column tubing to TUV: 0.004’’ ID 22.5’’ LG MP35N Welded Tube (p/n: 430002856), Assy, Pre-HRT to Column, Reusable MP35N (p/n: 700011809), ACQUITY UPLC™ 30 cm Column Heater/Cooler (p/n: 176015127) |
|
Column: |
Waters GTxResolve 250 Å Slalom Chromatography Column, MaxPeak™ Premier Technology, 2.5 µm, 4.6 x 300 mm and dsDNA 23K Ladder (p/n: 176006046) |
|
Mobile phase A: |
1X TAE buffer, pH 8.3 |
|
Column equilibration: |
Column conditioning was done by ramping the flow rate by 0.1 mL increments over 20 minutes to reach the final flow rate of 1.3 mL/min and further continued for another 40 minutes. |
|
Vials: |
Max Recovery Sample Vials (Waters p/n: 186009186 (vial) p/n: 186005827 (caps)) |
|
Column temperatures: |
40 °C |
|
Sample temperature: |
5 ± 3 °C |
|
Sample amount: |
1 µL |
|
Flow rate: |
1.3 mL/min |
|
Gradient: |
Isocratic |
|
Sampling rate: |
40 Hz |
Results and Discussion
Sensitive Detection of dsRNA
The capability of a GTxResolve 250 Å Slalom Chromatography Column to produce fast yet reproducible separations of large DNA molecules and plasmid topology analysis was demonstrated in a set of previous application notes.8-9 In the current study, the focus is on leveraging the GTxResolve 250 Å Slalom Chromatography Column for the sensitive detection of dsRNA. With advances in gene therapy, mRNA is playing an increasingly important role in therapeutic drug developments. mRNA molecules are synthesized by IVT reaction mediated by T7 RNA polymerase, which is known for its efficiency and high yield. However, this process often results in unwanted dsRNA by-products that contaminate the desired mRNA drug substance. As these dsRNA contaminants can trigger harmful immune responses, they are required to be minimized and documented in the final mRNA drug formulation.
Following PCR-based amplification of templates for sense and antisense RNA strands, IVT reaction was performed as described in the Experimental section. The synthesized sense and antisense strands were mixed and annealed to make dsRNA. These synthetic single stranded and dsRNA molecules served as model molecules for mRNA and dsRNA contaminants, respectively.
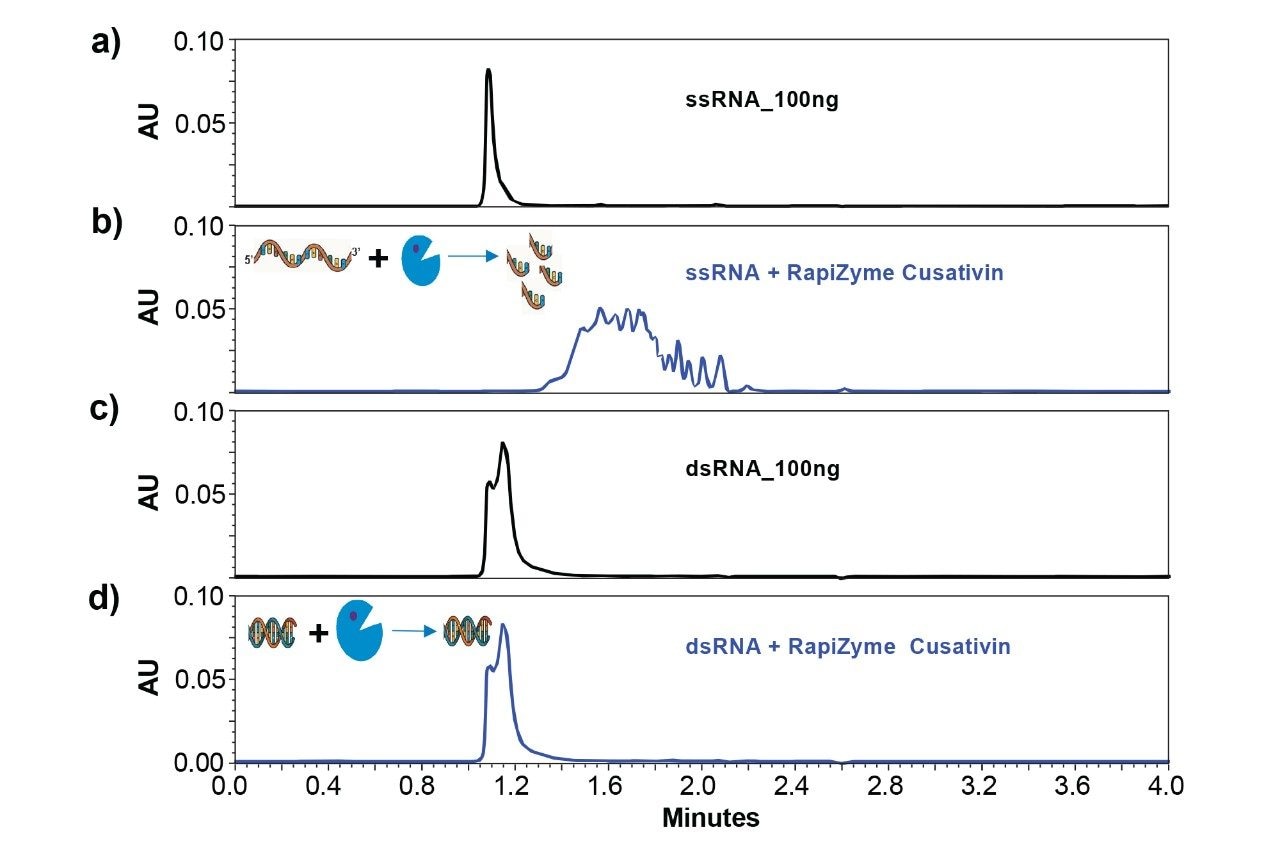
Figure 1 compares the detection of ssRNA and dsRNA (annealed form of sense and antisense strands) by slalom chromatography as well as agarose gel electrophoresis. Analysis of 3,000 nt ss and dsRNA (~100 ng) by GTxResolve 250Å Slalom Chromatography Column reveals a different peak profile for dsRNA (Figure 1, a(iii)) compared to ssRNA of either sense or antisense nature (Figure 1, a(i&ii). Overlay of these profiles suggests partial separation of dsRNA peak from ssRNA (Figure 1, a(iv). Although agarose gel electrophoresis could distinguish ss and dsRNA based on their relative migration (Figure 1b), >400 ng dsRNA (against 100 ng in slalom chromatography) was required for detection on an agarose gel.

Although dsRNA exhibited a different peak profile by slalom chromatography, only a small difference in peak retention time was noticed (Figure 1) compared to ssRNA. Therefore, to corroborate the true nature of annealed dsRNA, it was subjected to single strand-specific ribonuclease treatment. The previous unpublished studies with RapiZyme RNases indicated that while these enzymes efficiently cleave ssRNA, the dsRNA remained resistant to enzymatic cleavage. To verify the single stranded and double stranded nature of sense, antisense and annealed forms of the RNA analytes, the samples of RapiZyme Cusativin treatment (see methods) followed by slalom chromatography were subjected. Figure 2 shows the combined effect of ribonuclease treatment and slalom chromatography on these RNA samples. While the ssRNA (either sense or antisense) was efficiently degraded by cusativin treatment (Figure 2a vs 2b), the annealed RNA sample remained intact even after 1 hour exposure to the RapiZyme Cusativin while maintaining identical peak profile (Figure 2c vs 2d). This observation confirms the double stranded nature of the annealed RNA sample. It may be noted that the digestion products of ssRNA exhibit multiple peaks beyond the intact RNA presumably due to their entry into the stationary phase pores and subsequent separation by hydrodynamic chromatography.
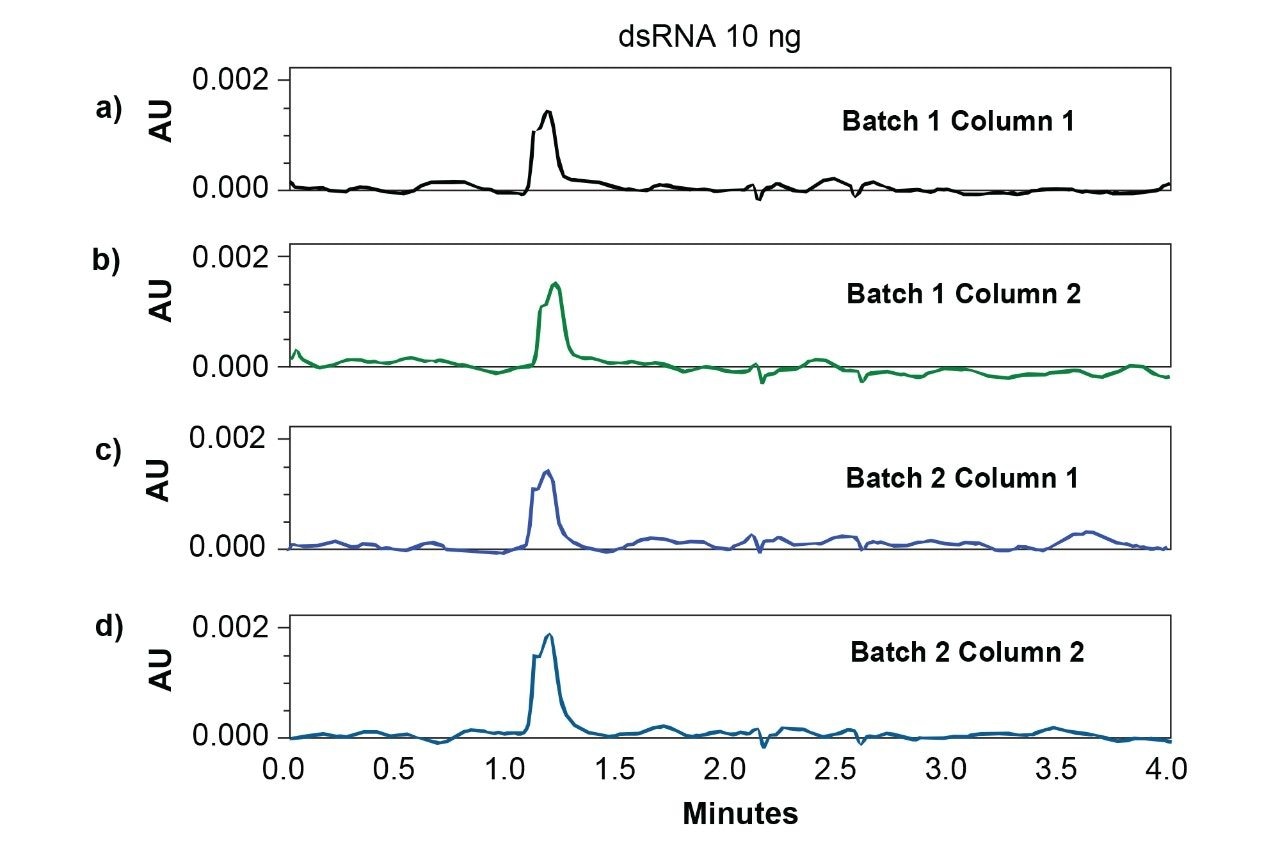
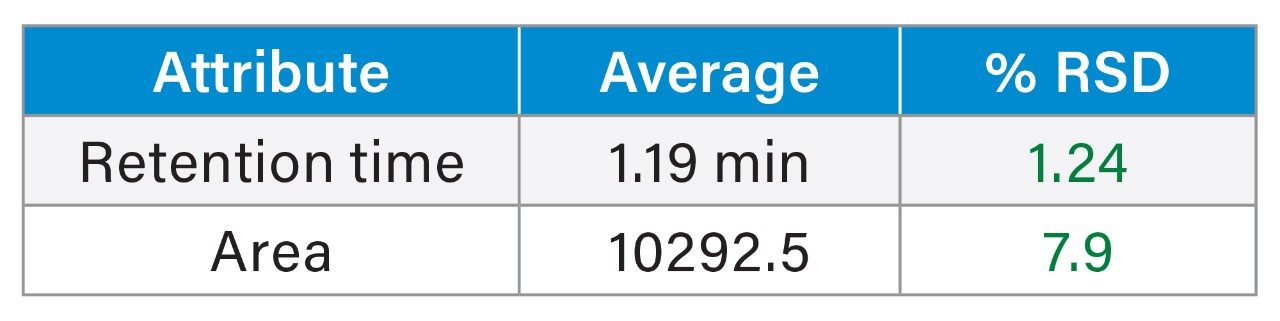
To evaluate the column-to-column and batch-to-batch reproducibility of slalom separations at lowest levels of detection, 3 kb dsRNA samples were analyzed by using Waters GTxResolve 250 Å Slalom Columns packed from two different batches of packing material. As little as 10 ng of dsRNA was reproducibly detected on multiple columns and batch materials (Figure 3). This is >40X improvement in detection compared to agarose gel electrophoresis. Moreover, these columns made from multiple batches also demonstrated outstanding reproducibility, as evidenced by the exceptionally low percent relative standard deviations (%RSD) for various peak parameters including retention times and peak area (Table 1).
Conclusion
This study demonstrates the exceptional sensitivity and reproducibility of the Waters GTxResolve 250 Å Slalom Chromatography Column for the detection and analysis of dsRNA impurities in mRNA samples. Although this work was carried on a model ss and dsRNA system, it is believed that this approach will be more broadly applicable to assaying IVT reactions for dsRNA. In future work, the potential interference of self-folded conformational states from the mRNA drug substance itself will also be investigated.
As reported here, slalom chromatography offers a 40-fold improvement in sensitivity over agarose gel electrophoresis, which enables the identification of dsRNA contaminants with as little as 10 ng of impurity. The column also showed outstanding reproducibility across multiple batches, reinforcing its reliability for routine analytical workflows.
- A 40-fold more sensitive detection of the dsRNA in <6 minutes compared to 60 minutes agarose gel electrophoresis.
- Precise discrimination between ssRNA and dsRNA by the integration of RapiZyme Cusativin-based enzymatic treatment with slalom chromatography. Cusativin selectively digested ssRNA while leaving dsRNA intact, allowing clear chromatographic differentiation.
Overall, the combined use of RapiZyme RNase and GTxResolve Slalom Columns provides a robust, sensitive, and reproducible platform for the detection, quantification, and characterization of dsRNA impurities so that safe and effective mRNA-based therapeutics can be enabled.
References
- Kang DD, Li H, Dong Y. (2023) Advancements of in vitro transcribed mRNA (IVT mRNA) to enable translation into the clinics. Adv Drug Deliv Rev. 199:114961. https://doi.org/10.1016/j.addr.2023.114961
- Gholamalipour Y. et al. (2018) 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses, Nucleic Acids Research, 46: 9253–9263. https://doi.org/10.1093/nar/gky796
- Liu X., et al. (2025). Research progress on immune mechanism and control strategy of dsRNA impurities in mRNA vaccine. Expert Review of Vaccines, 24(1), 457–469. https://doi.org/10.1080/14760584.2025.2510335
- Liu J. et al. (2024) An improved method for the detection of double-stranded RNA suitable for quality control of mRNA vaccines. Protein Cell. 15(11):791–795. https://doi.org/10.1093/procel/pwae043
- Karikó K. et al. (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 39(21):e142. https://doi.org/10.1093/nar/gkr695.
- Clark N.E. et al. (2024) An immuno-northern technique to measure the size of dsRNA byproducts in in vitro transcribed RNA. Electrophoresis. 45:1546–1554. https://doi.org/10.1002/elps.202400036
- Gritti, F. (2025) Retention mechanism in slalom chromatography: Perspectives on the characterization of large DNA and RNA biopolymers in cell and gene therapy. J Chrom A 1743, 465691 (2025) https://doi.org/10.1016/j.chroma.2025.465691
- Vaishnav, J. et al. (2025) A New Alternative to Gel Electrophoresis: Higher Resolution and Faster Analysis of Large Nucleic Acids by Rigorously Designed GTxResolveTM 250Å Slalom Columns. Waters Application Note: 720008921
- Vaishnav, J. et al. (2025) Plasmid Topology and Digest Analysis Using a GTxResolve™ 250 Å Slalom Chromatography Column. Waters Application Note: 720008954
- B. Addepalli et al. (2024) Tunable Digestions of RNA Using RapiZyme™ RNases to Confirm Sequence and Map Modifications. Waters Application Note: 720008539
720009005, August 2025