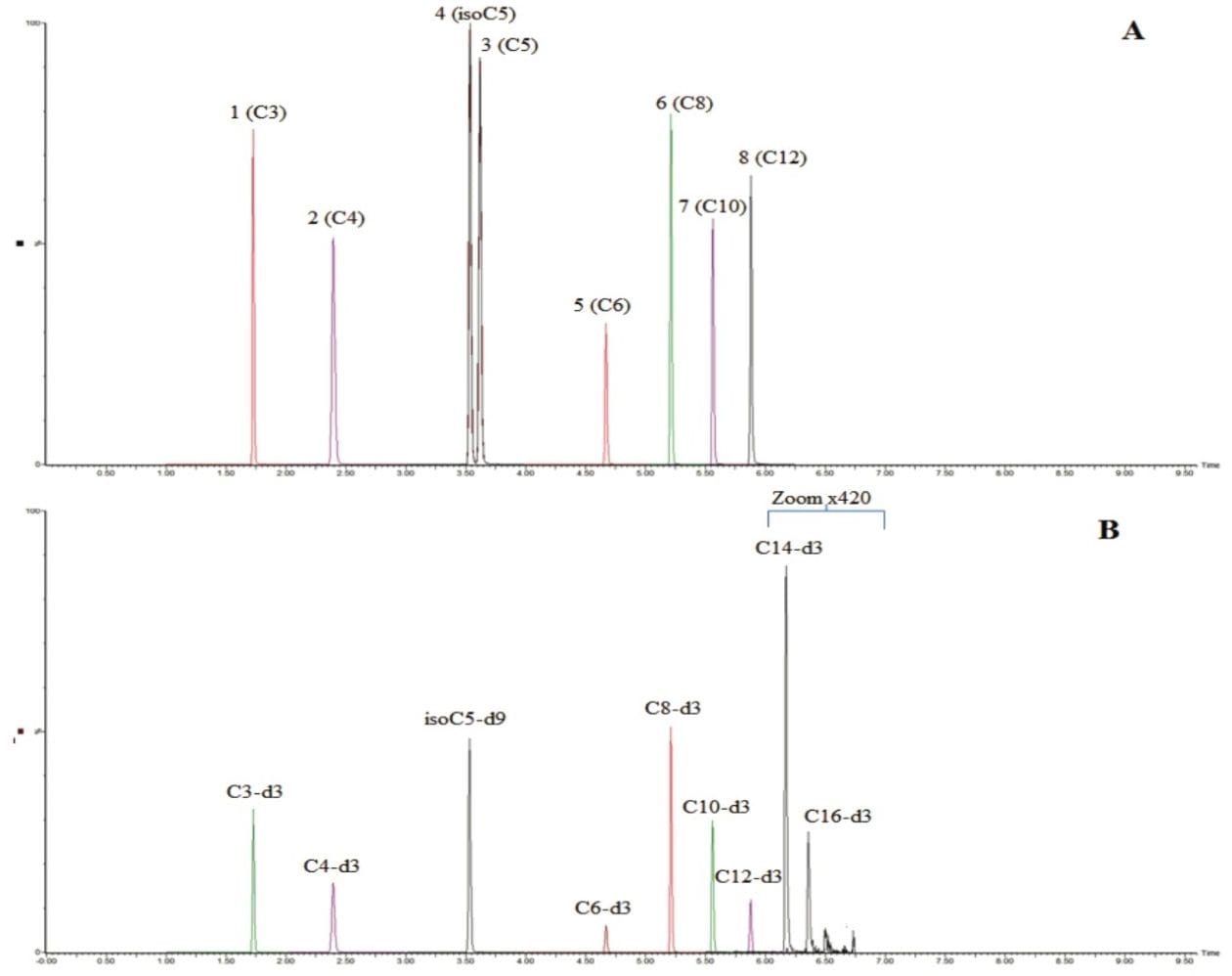
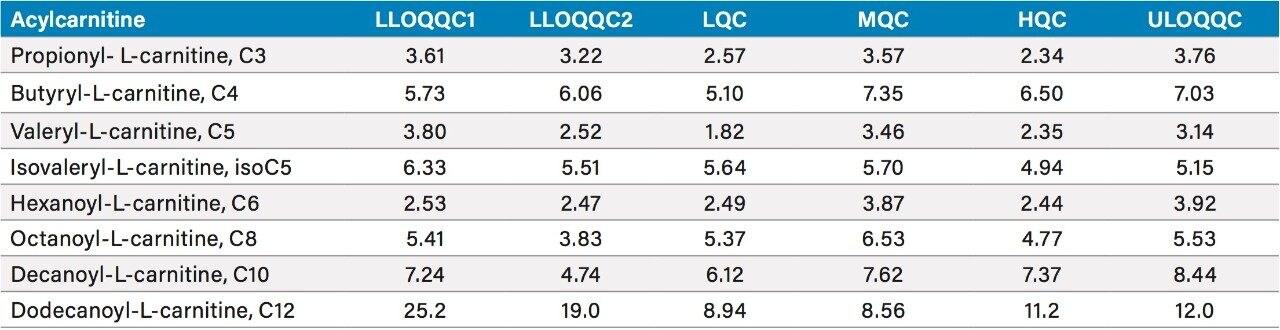
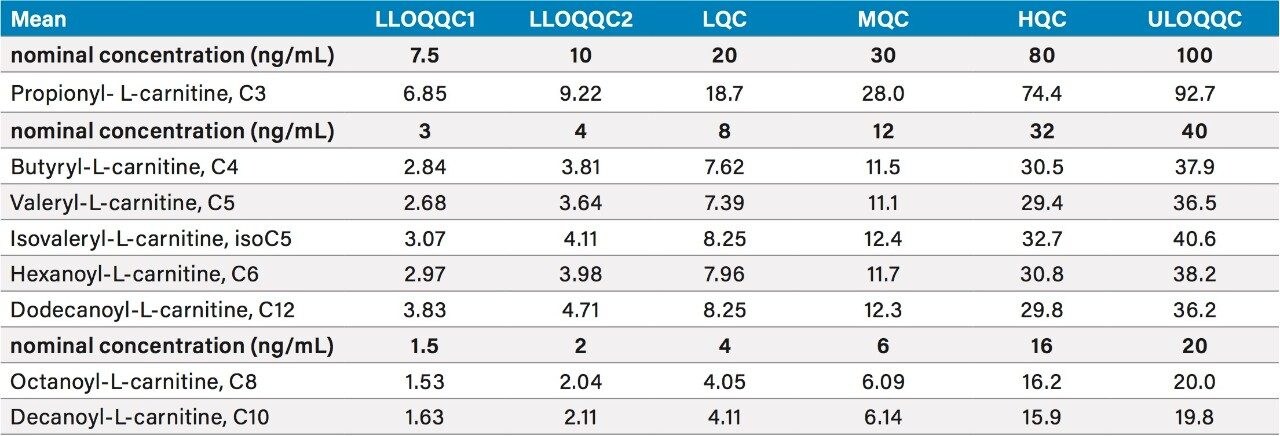
The chromatographic separation was developed to provide sufficient resolution of the analytes, with a run time of 10 minutes consistent with medium to high throughput analysis, while maintaining the separation between the two pairs of isomeric species isovaleryl-L-carnitine (isoC5) and valeryl-L-carnitine (C5), and isobutyryl-L-carnitine (isoC4) and butyryl-L-carnitine (C4). Due to their relatively low concentrations in urine, compared to carnitine itself and some other high concentration acylcarnitines, the analytes measured in this method, propionyl-L-carnitine (C3), butyryl-L-carnitine (C4), isovaleryl-L-carnitine (isoC5), valeryl-L-carnitine (C5), hexanoyl-L-carnitine (C6), octanoyl-L-carnitine (C8), decanoyl-L-carnitine (C10) and dodecanoyl L-carnitine (C12) need to be quantified using a method optimized for low concentration acylcarnitines. Dodecanoyl L-carnitine (C12), tetradecanoyl-L-carnitine (C14) and hexadecanoyl-L-carnitine (C16) are too low in concentration in the control urine to be quantified using the sample preparation method for this analysis, however, the internal standards are displayed in Figure 1B for the purposes of demonstrating retention time and could allow for testing of these analytes.