The samples analyzed consisted of tryptically digested, undepleted human plasma from three different conditions spiked with Biognosys PQ500 SIL peptides. Sample groups consisted of controls (n=6), chronic obstructive pulmonary disease (n=6), and asthma (n=6). Individual samples per group were pooled to provide three working samples. In addition, a pool of all conditions/samples acted as a QC. An ACQUITY UPLC I-Class System was equipped with an ACQUITY UPLC CSH130 C18, 1 mm x 100 mm long analytical column (p/n 186006934). Samples were analyzed in triplicate (QC = 5 injections) at two different loadings of 5 ug and 10 ug, which incorporated the PQ500 at ‘Injection Equivalents’ of one and two respectively. The samples were separated using a reversed phase gradient from 1% to 40% acetonitrile (+0.1% formic acid) over 15, 30, or 45 minutes at a flow rate of 50 µL/min. In order to minimize any post column peak broadening, a short piece of 60 micron I.D. PEEK tubing connected the output of the column to a low flow ESI probe. The mass spectrometer acquisition mode employed was SONAR DIA, which has been described elsewhere.² The same conditions were applied here. Data were processed using Progenesis QI for Proteomics and Spectronaut Pulsar X (Biognosys, Schlieren, Switzerland). Searches were performed using carbamidomethyl C (fixed) and oxidation of methionine (variable) modifications in addition to a 1% FDR.
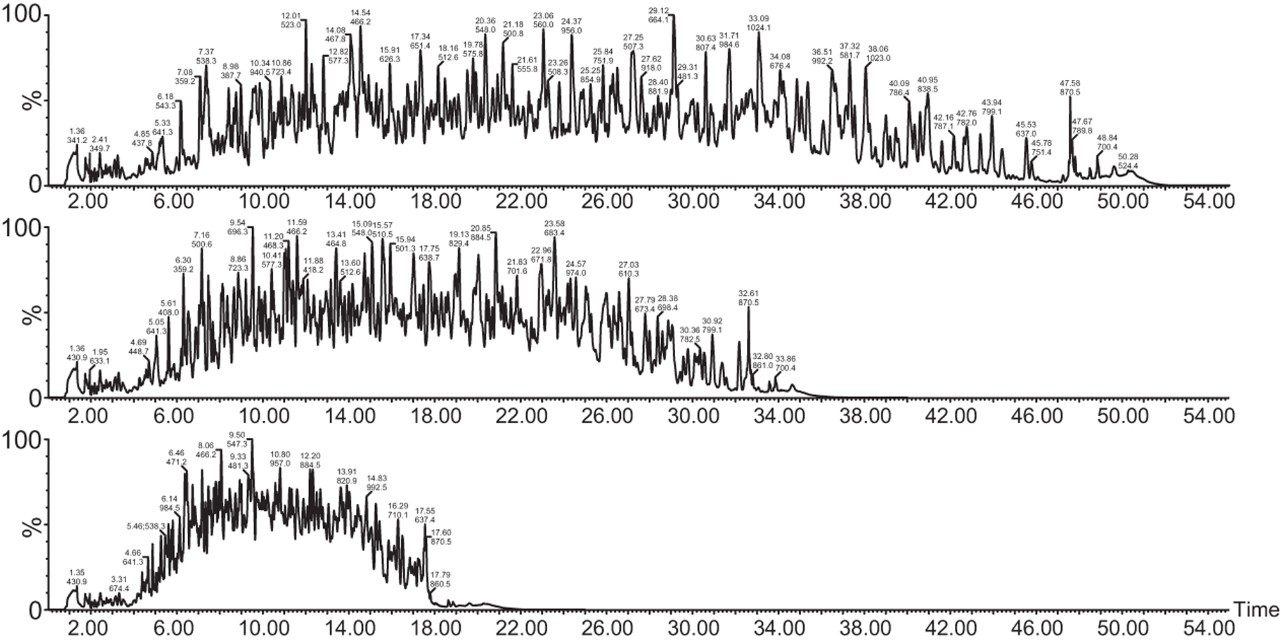
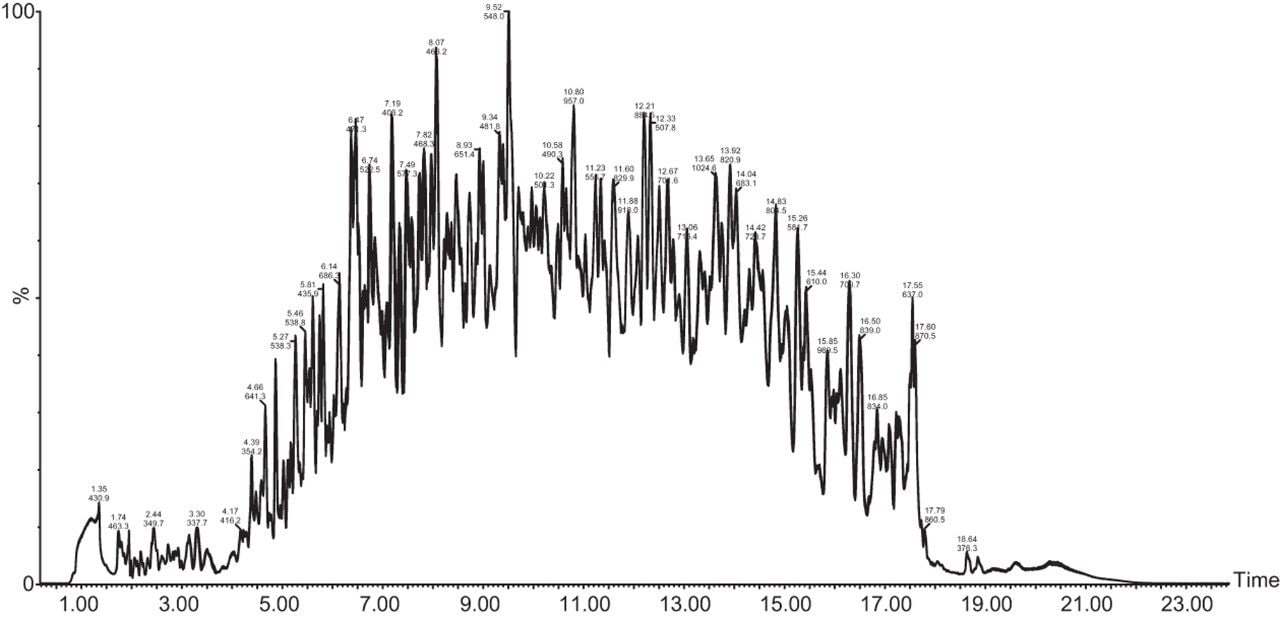
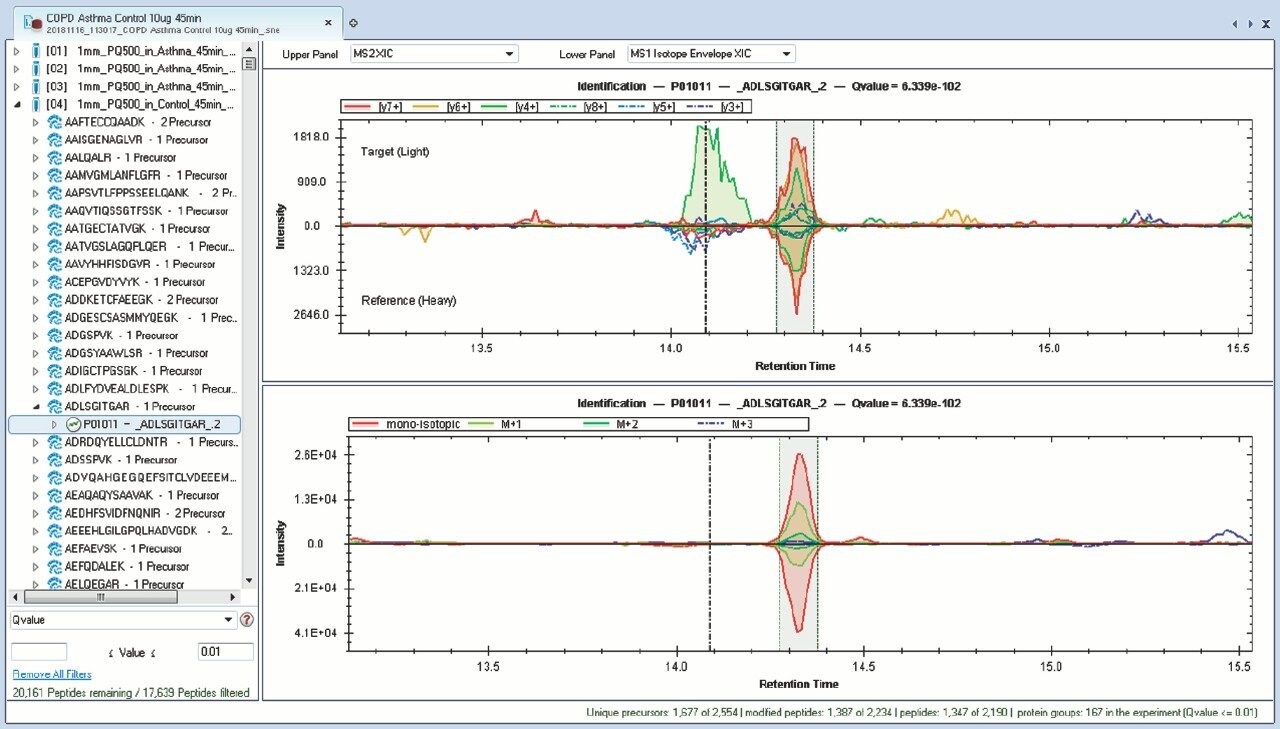
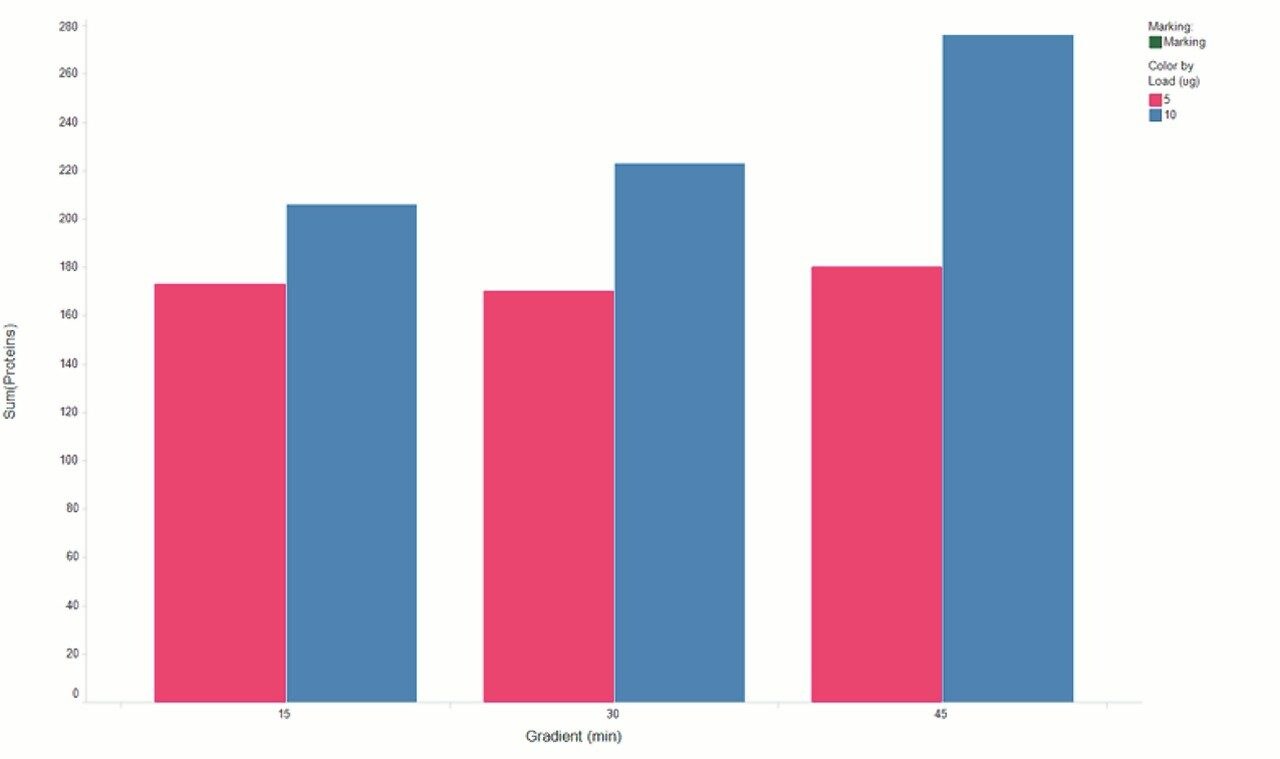
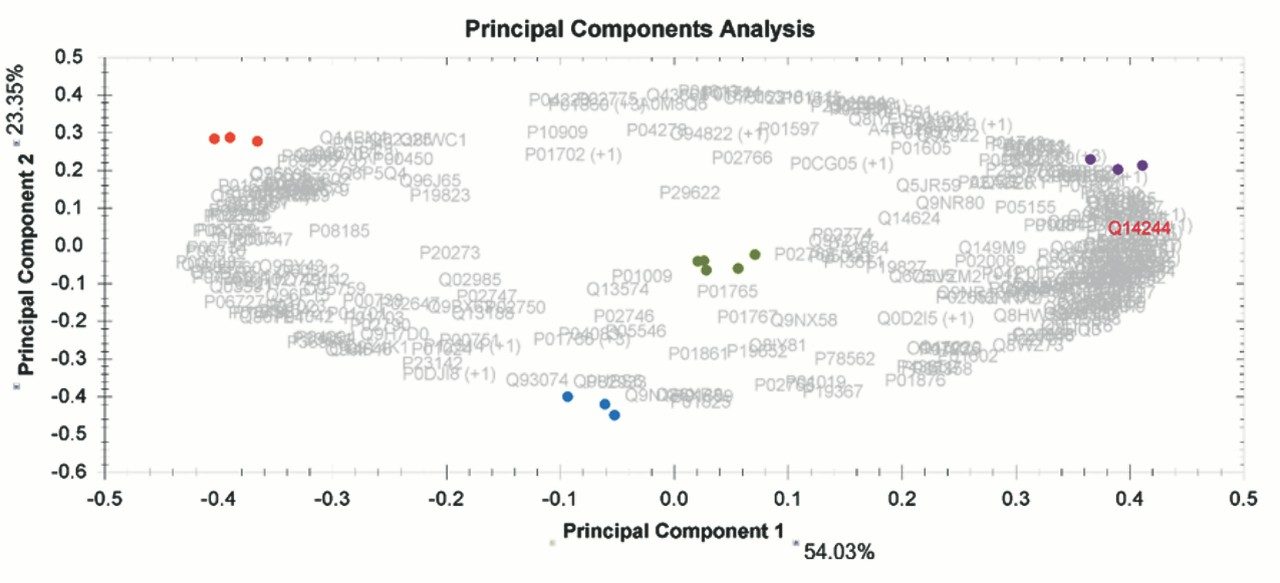
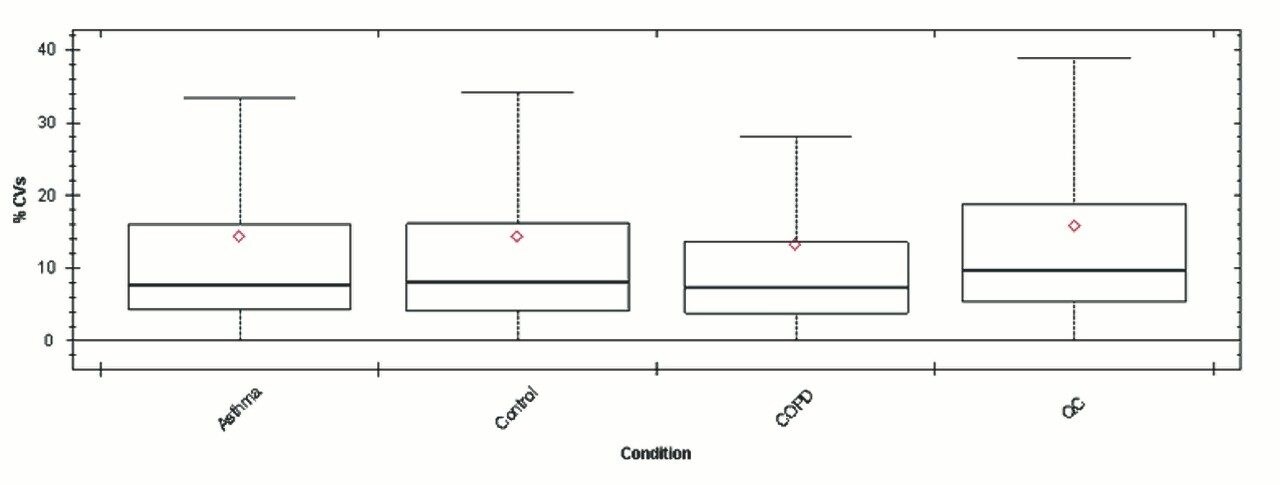
Chromatograms in Figure 1(a) represent the effect of gradient length on peptide separation, where shown are data from the three different gradient lengths and a load of 10 ug. The excellent chromatographic reproducibility obtained for the shortest gradient length is shown in Figure 1(b), which represents three overlaid chromatograms for 15 min gradient length and a 10 ug load. When data is processed using Spectronaut, heavy and light analogues are identified from the peptide panel. For a 10 ug load and 45 minute gradient, one such heavy and light identification is shown in Figure 2 for the peptide EDVYVVGTVLR from C4b-binding protein alpha chain (Uniprot accession: P04003). In total, 159 light/heavy pairs are identified in this experiment. The total number of quantified protein identifications from searches performed within Progenesis QI for Proteomics are shown in Figure 3. Gradient length had little effect on protein IDs for the 5 ug loadings but longer gradients were more beneficial for the 10 ug loading. The samples used for this study consisted of two different respiratory disease states, normal control, and a pooled QC. The PCA plot shown in Figure 4 indicates that separation of the different groups is readily achieved and corresponding CVs of <8% (Figure 5) further demonstrate the technical reproducibility of the data.