The analyzed samples consisted of tryptically digested, undepleted human plasma from three different conditions. Sample groups consisted of controls (n=6), chronic obstructive pulmonary disease (n=6), and asthma (n=6). Individual samples per group were pooled to provide three working samples.
Peptides were separated using an ACQUITY UPLC M-Class System, which was equipped with an ACQUITY UPLC Peptide CSH C18, 300 µm I.D. × 100 mm long analytical column. Samples were analyzed in triplicate (QC=5 injections) based on 5 µg loadings. The samples were separated using a reversed-phase gradient from 1 to 40% acetonitrile (+0.1% formic acid) over 45 minutes at a flow rate of 50 µL/min. Data were collected using a SYNAPT XS which operated using the SONAR mode of acquisition. Data were processed using Progenesis QI for Proteomics and database searched against a human database, consisting of UniProt reviewed sequences. Searches were performed using carbamidomethyl C (fixed) and oxidation of methionine (variable) modifications in addition to a 1% FDR.
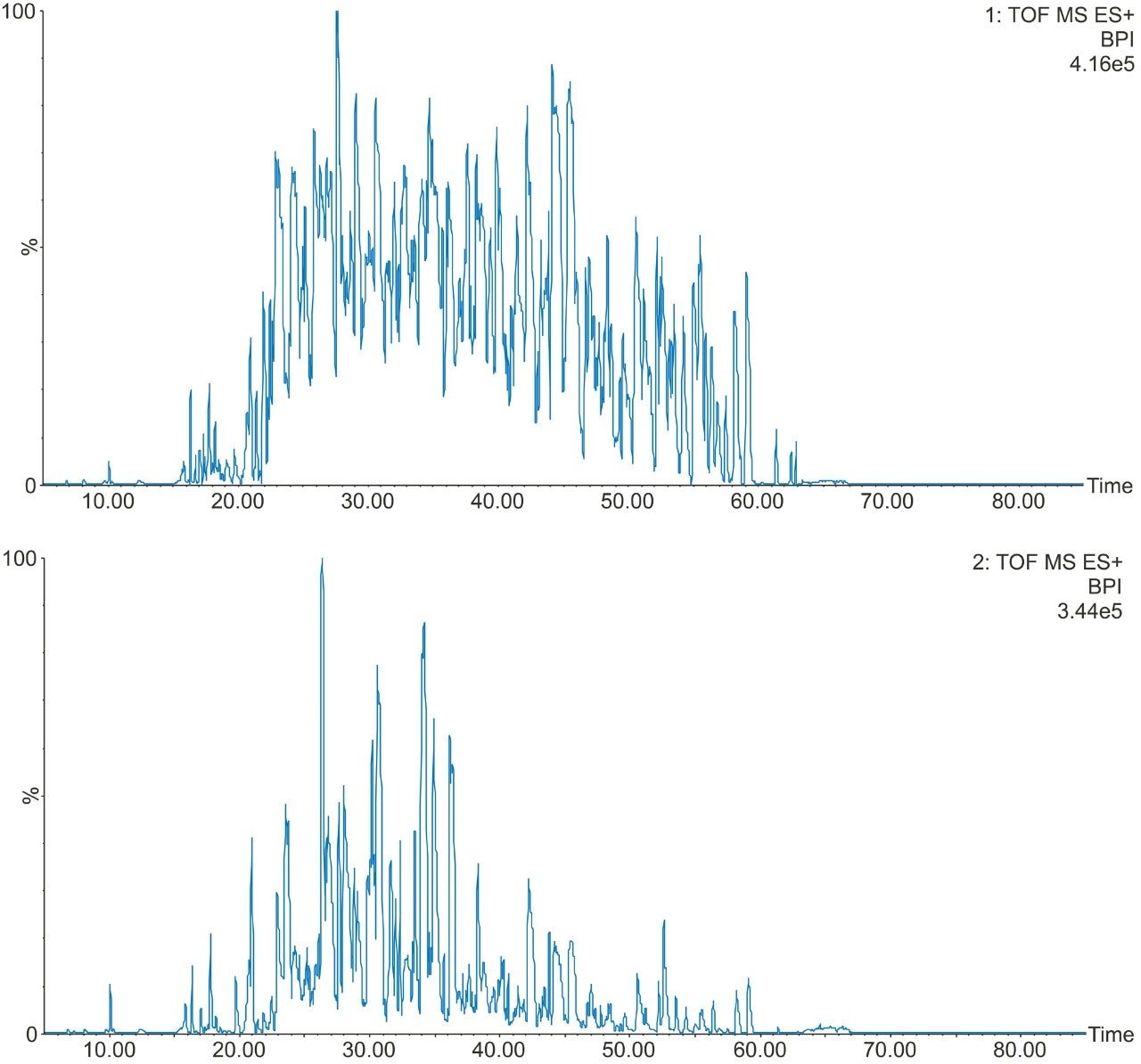
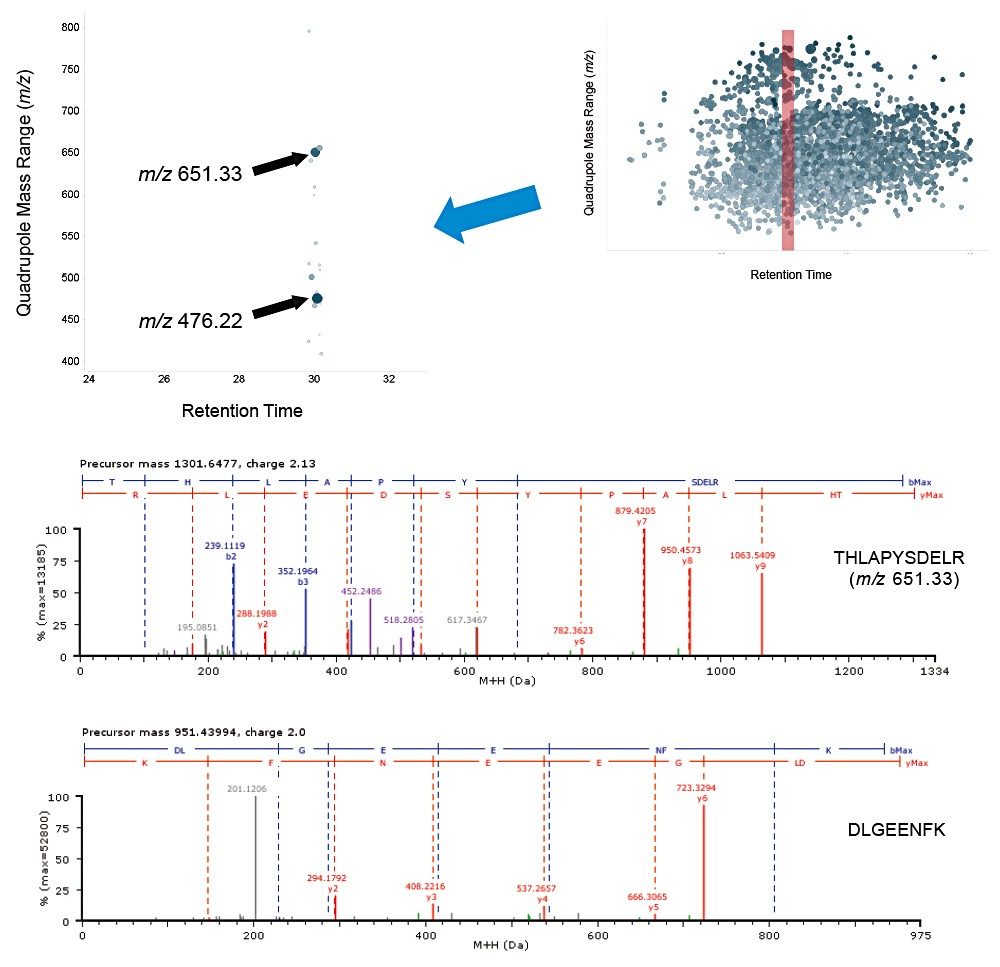
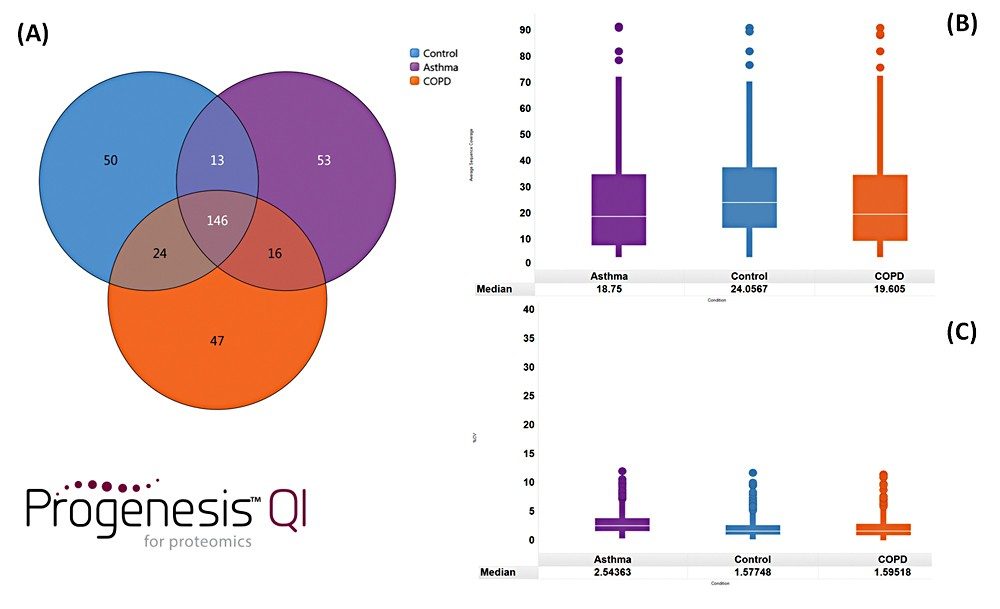
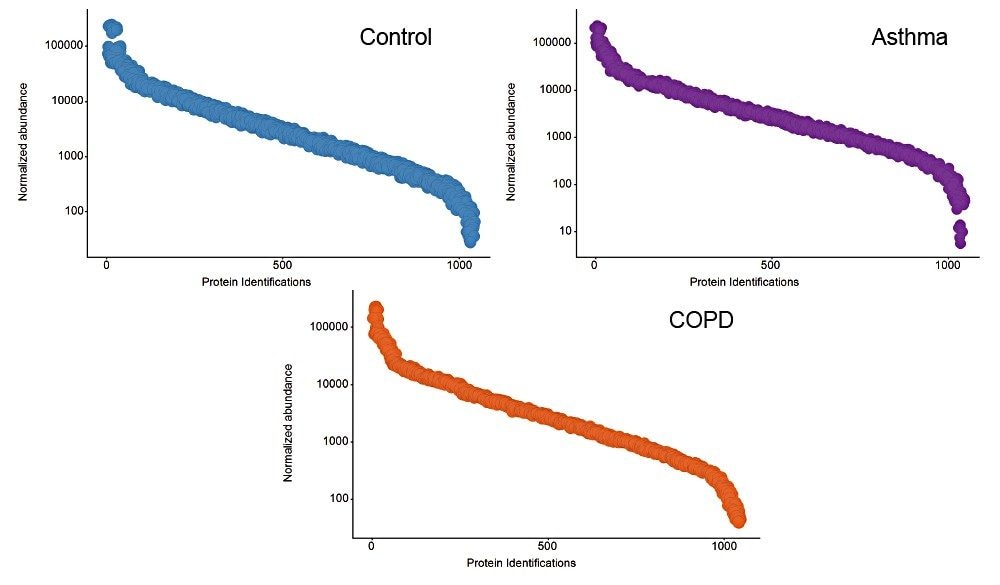
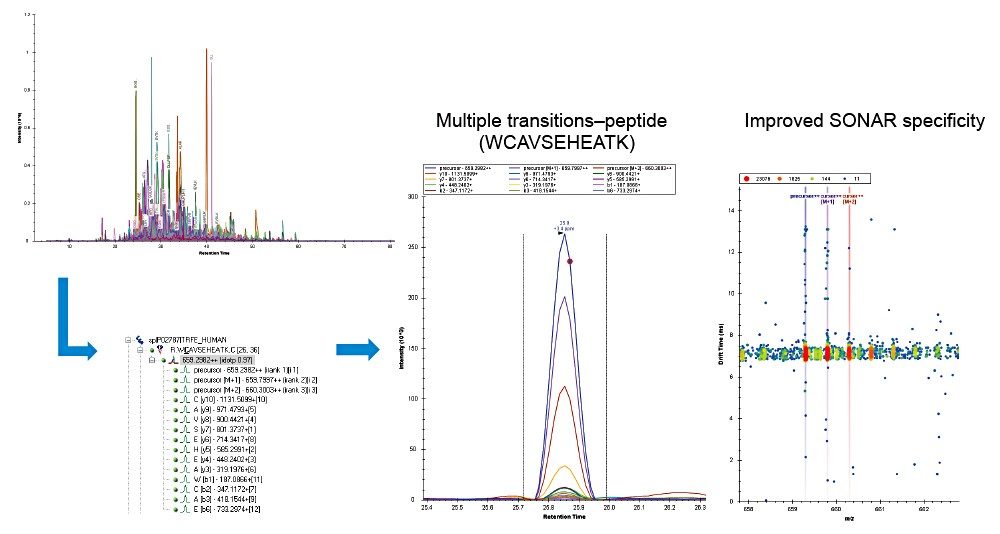
Example chromatograms representing SONAR acquisition are shown in Figure 1 for both precursor and fragment ion data. The benefits of high selectivity when implementing SONAR have previously been reported.1,2 Figure 2 shows the enhanced specificity provided by the scanning quadrupole implemented for increased peptide identification. Data processing with Progenesis QI for Proteomics identified a total of 349 proteins with the majority of identifications overlapping between all three cohorts (Figure 3A), whilst the median sequence coverage achieved varied between 18–25% across groups (Figure 3B). The ability to reproducibly identify and quantify analytes of interest is important to ensure consistency over the analysis. Figure 3C shows the coefficient of variation (CV) for the normalized abundance across technical replicates for each condition, with a median CV less than 3% being achieved across each condition. Furthermore, a linear dynamic range of 3–4 orders magnitude is demonstrated for each condition (Figure 4).