This study shows how CCS areas can be used as an additional point of reference to add confidence to the correlation between a MS imaging data set (collected using MALDI), and the subsequent molecular identifications by MS-MS.
The ion mobility cell of the SYNAPT G2-Si HDMS Mass Spectrometer can be easily calibrated via IntelliStart which automatically calculates the necessary calibration constants. For the calibration of the system poly-alanine was deposited on a MALDI target plate, mixed with the negative mode matrix 9-aminoacridine (9-AA).
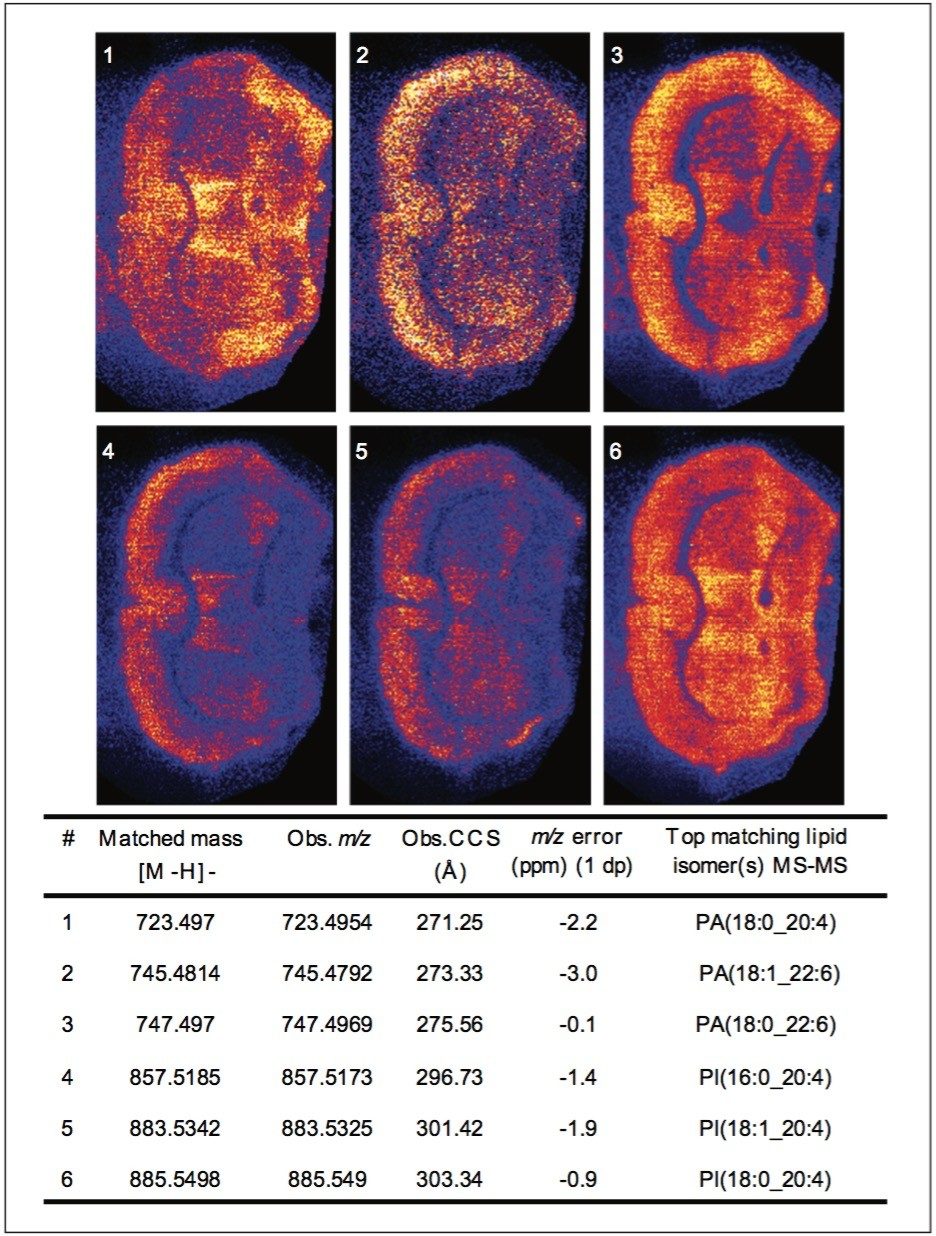
For the MS imaging experiment, a thin section of mouse brain was produced using a cryotome and deposited on a non-conductive glass slide. The 9-AA matrix was applied evenly to the sample by spray coating. The MS imaging data was acquired at a spatial resolution of 45 μm in negative ion mode across a mass range of m/z 50-1,150, utilising TriWave ion guide optics. This acquisition mode can separate ions according to their ionic mobility in the gas phase. To maintain high accurate mass performance, an external lock mass was used. The MS imaging dataset was subsequently processed using High Definition Imaging (HDI) Software v1.3 for detailed image analysis. Lipid peaks present in the imaging data were compared to the Lipid Maps data base (www.lipidmaps.org). In this case, 168 lipid candidates were identified based on mass accuracy better than +/-3 ppm. The CCS areas of the lipid candidates were calculated using the polyalanine calibration constants.
Lipids were extracted from two consecutive tissue sections by carefully depositing droplets of a solvent mixture onto the tissue section. Then, the extracted lipids were withdrawn by removing the extraction solvent. A 2:1 chloroform:ethanol solvent mixture was used for lipid extraction on one of the tissue section, while 4:1 ethanol:water was used on the second serial tissue section to be consistent with the solvent mixture used in the MS imaging experiment. It also offered a more complete picture of the lipids characterizing the tissue section.
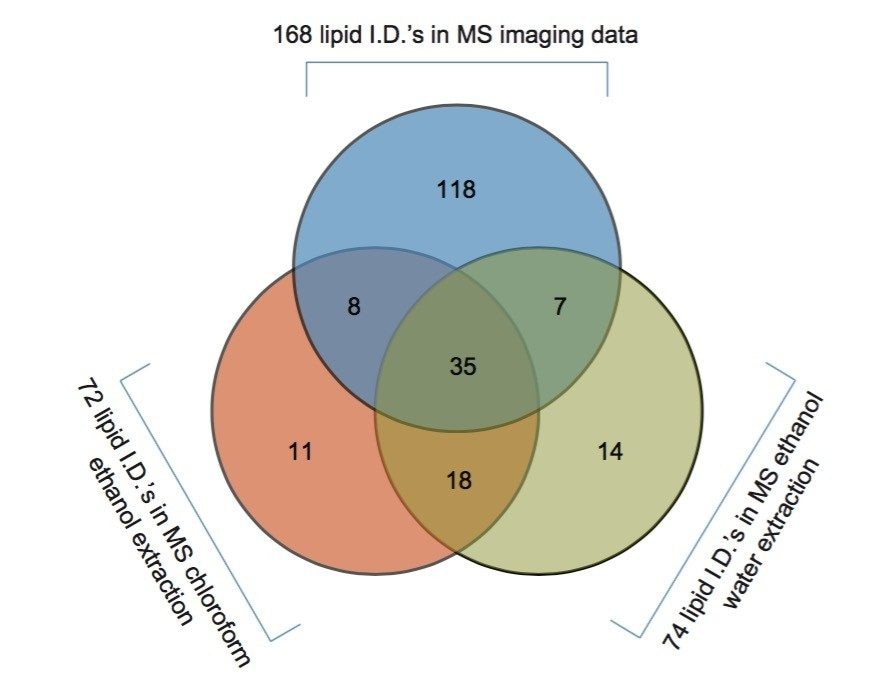
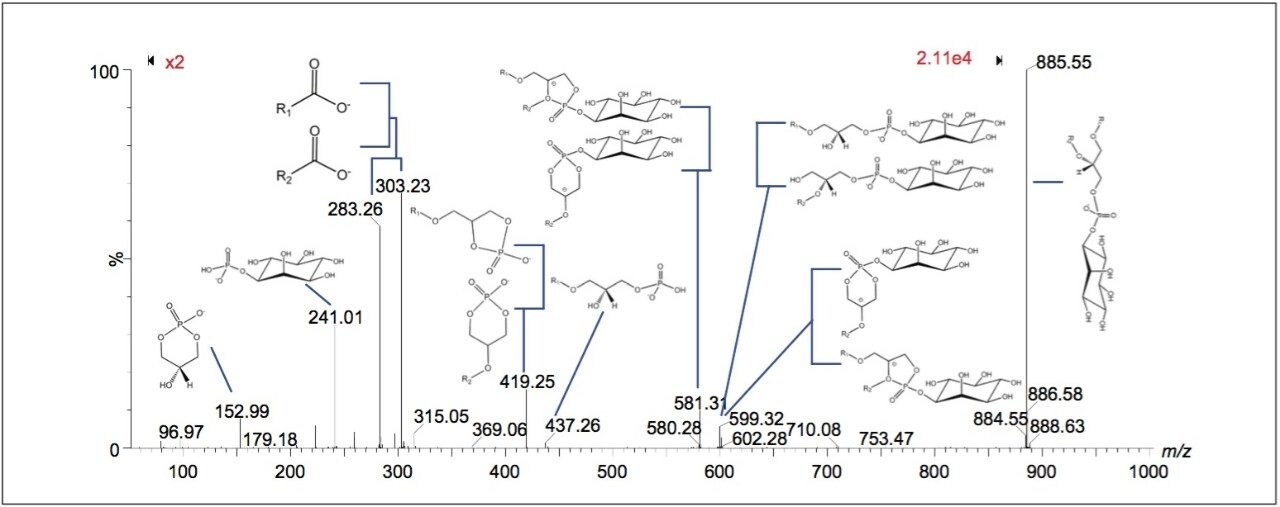
The two lipid extracts were spotted separately onto a standard MALDI target plate using 9-AA as the matrix. After MS analysis of each lipid extract, the peaks present were compared to the lipid maps database and the CCS areas calculated. The resulting lipid candidate lists were then compared to the list of 168 lipid candidates from the MALDI MS imaging dataset. The CCS areas of the peaks were cross validated (+/-0.5%) between the three datasets. The average CCS difference for the matches was found to be +/- 0.11%. 50 candidate lipid peaks were identified as being common to the MALDI imaging data set and one of the two lipid extract MS datasets (Figure 1).