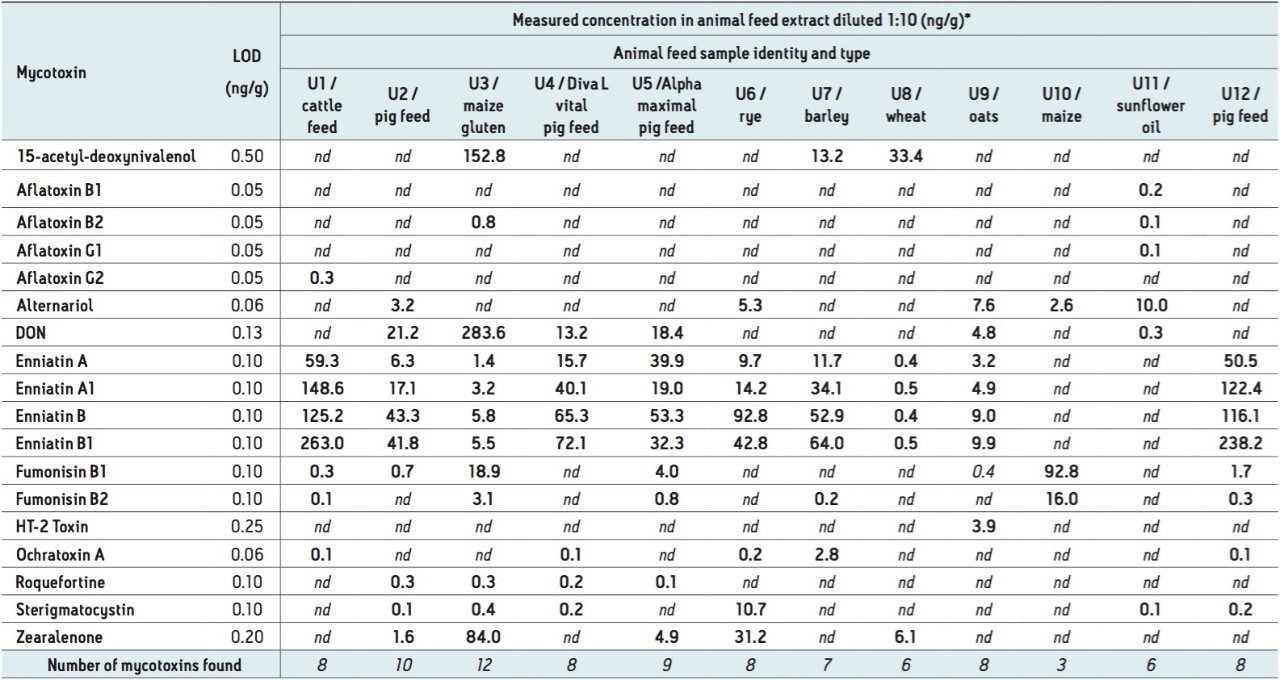
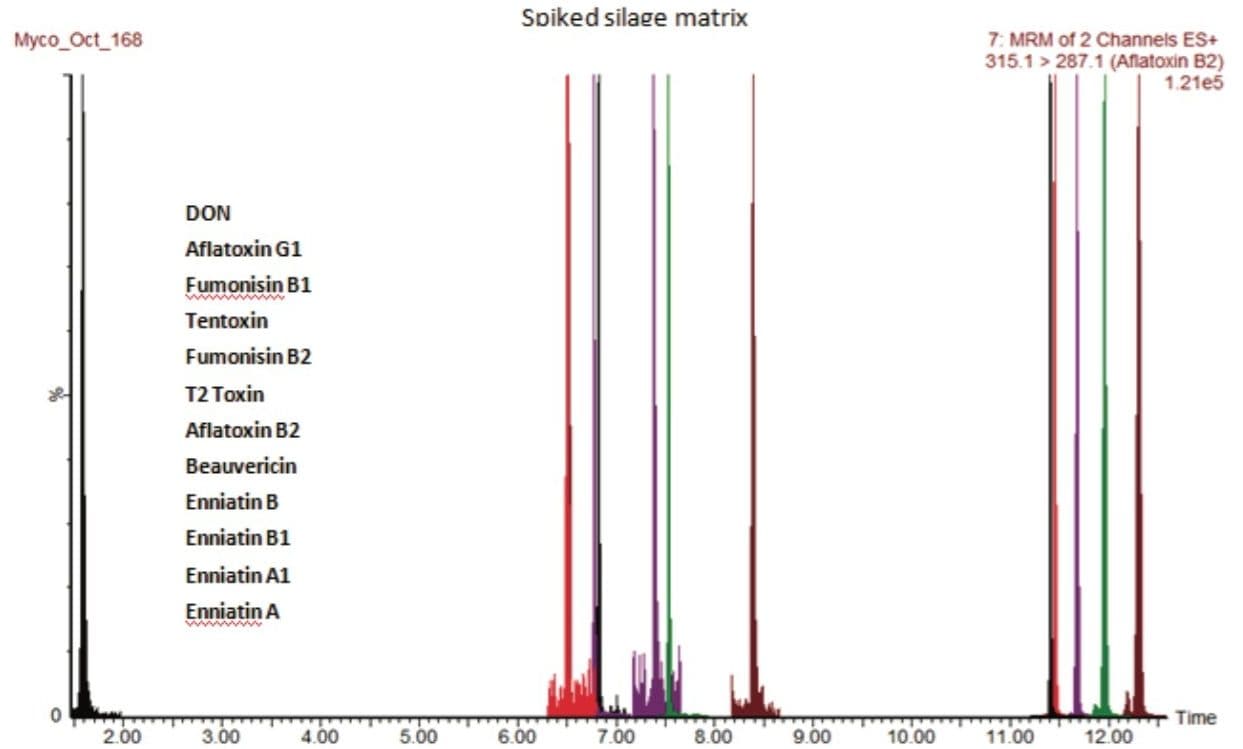
There are now over 400 recognized mycotoxins that may be found in animal feedings materials and it has been reported that as much as 25% of the world’s cereal grains may be contaminated with mycotoxins.1
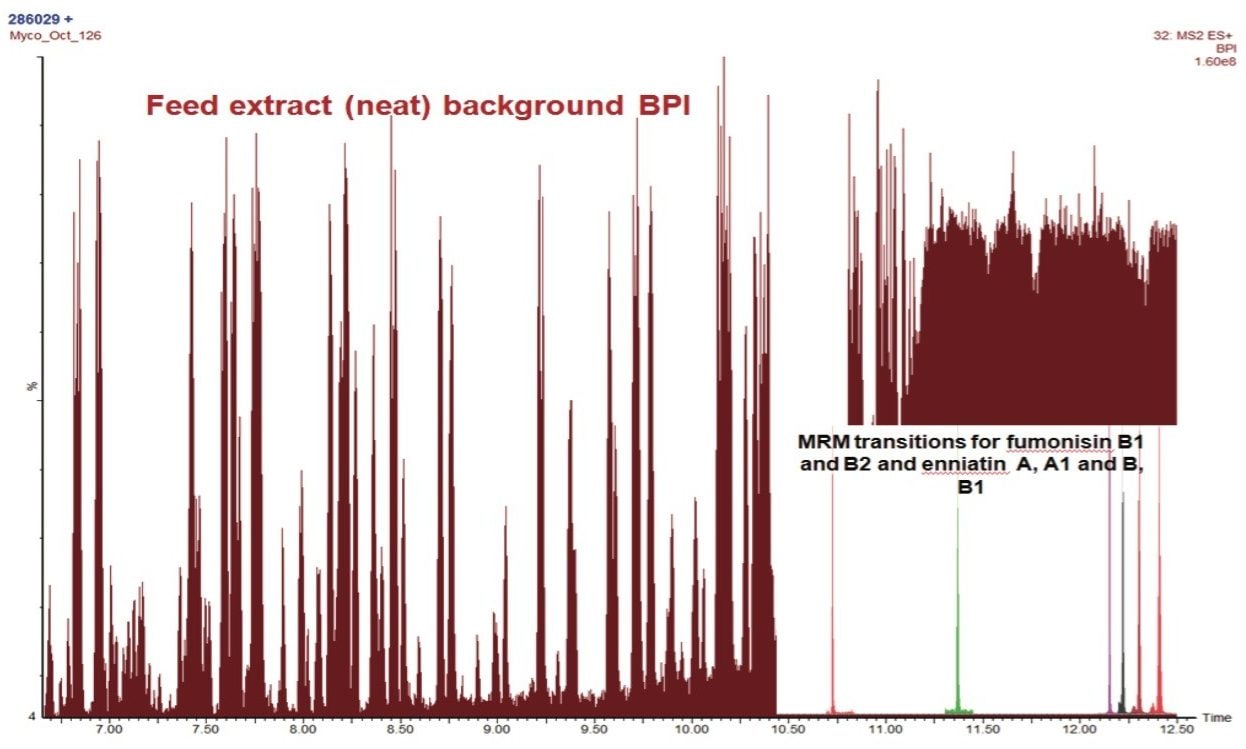
The analysis of animal feedingstuffs including silage represents a major technical challenge due to the complexity and in-homogeneity of these matrices. Although permitted limits for mycotoxins are set at relatively high (µg kg-1) concentrations in the EU,2,3 toxic effects such as immunotoxicity and feed uptake problems in certain species (poultry and porcine) are often observed at sub μg kg-1 concentrations.4 For this reason there is often a requirement to achieve low detection limits in feedingstuffs. There is also a potential for co-contamination due to pre- and post- harvest infestation resulting in the occurrence of tricothecenes, beauvercin and enniatins, fumonisins, ochratoxin, T2, HT-2, and alternaria toxins for example within a single feed sample.5
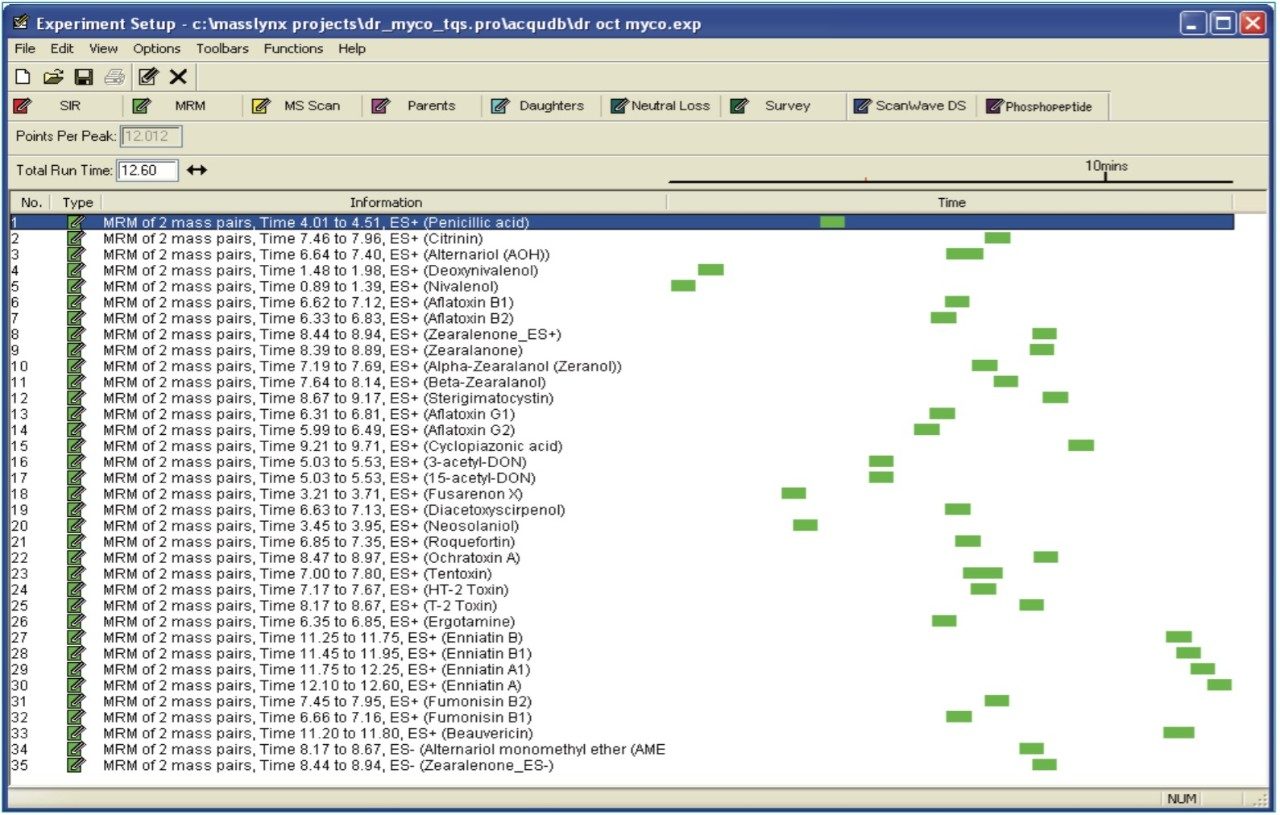
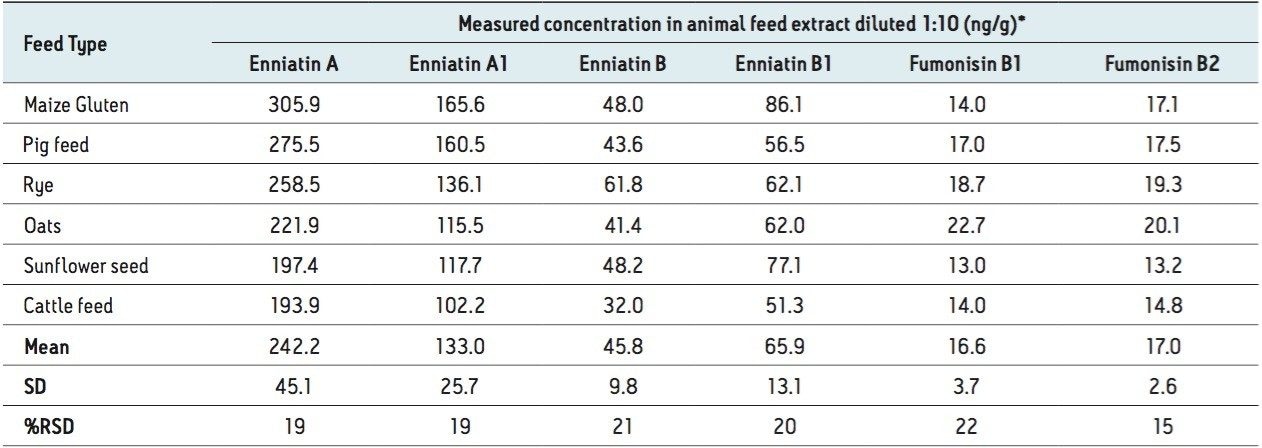
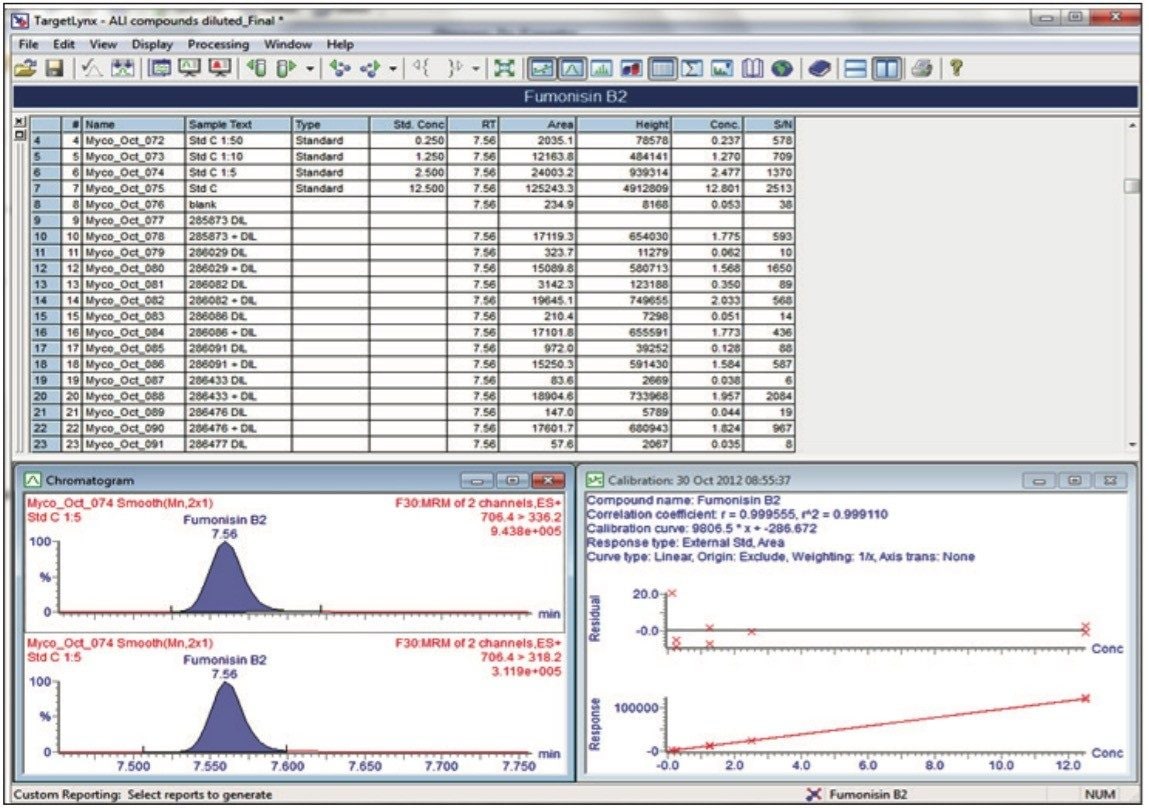
In this application note, we report the development of a quantitative method for the determination of 33 relevant mycotoxins in a variety of animal feed and silage extracts. A Waters ACQUITY UPLC I-Class System coupled to a Xevo TQ-S was used for rapid, high quality, and ultra-sensitive analysis of multiple mycotoxins in feed extract. Our goal was to investigate the effect of matrix dilution and enhanced instrument sensitivity to overcome common analytical challenges such as ion suppression and to reduce the effects of matrix variability.