MRM transitions were designed iteratively starting with in silico prediction, followed by experimental validation and comprehensive MRM testing of all of the candidate peptides. In short, Cys, Met, and RP/KP containing peptides were excluded, as well as missed cleaved and modified peptides. The minimum and maximum amino acid lengths were 8 and 25 amino acids, respectively. VerifyE Software was employed as a filter to exclude non-proteotypic, interfered transitions using 2 Th precursor and product ion tolerances and a 2-min retention time window. A pooled sample comprising equal amounts of all patient and control subject samples was used to examine the usability of the predicted final transitions in terms of response and interference. The retained transitions were manually inspected and curated using TargetLynx, and analyzed and scored with mProphet.
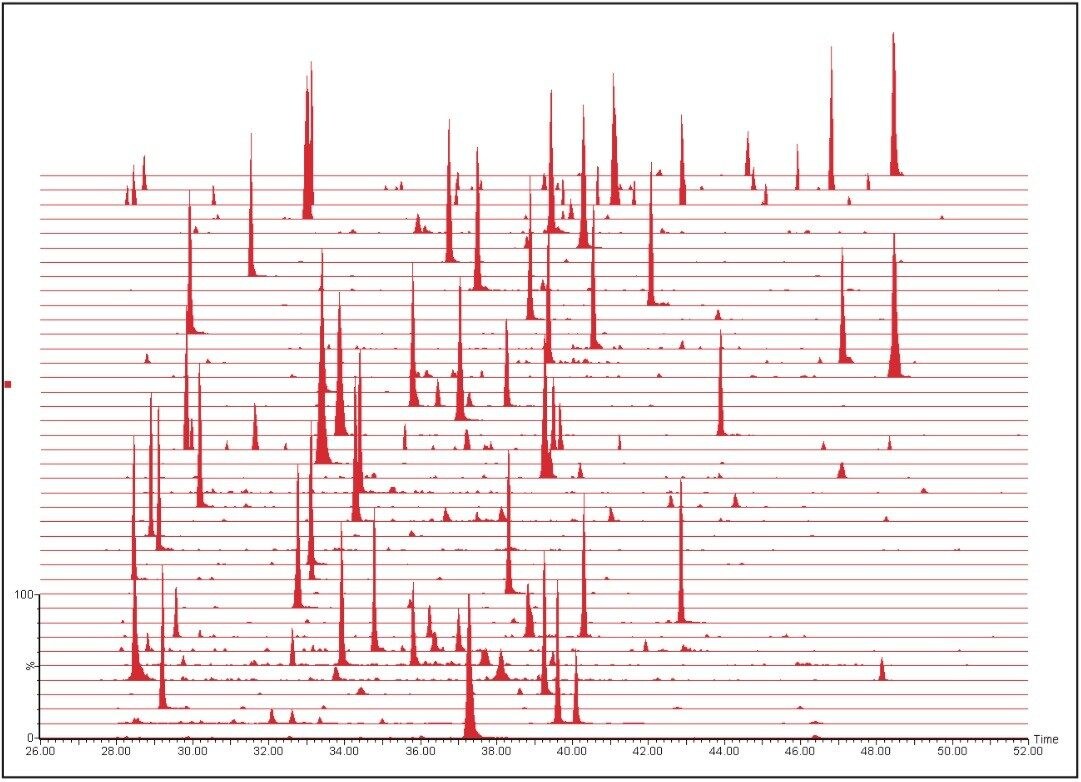
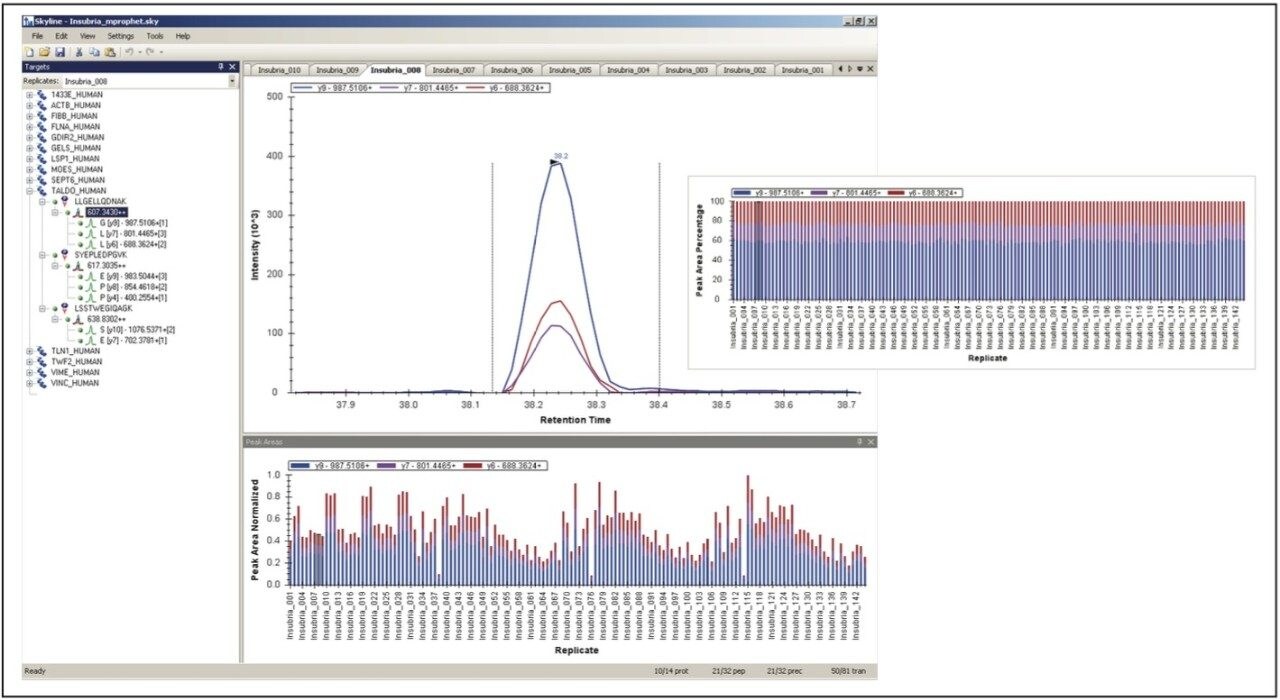
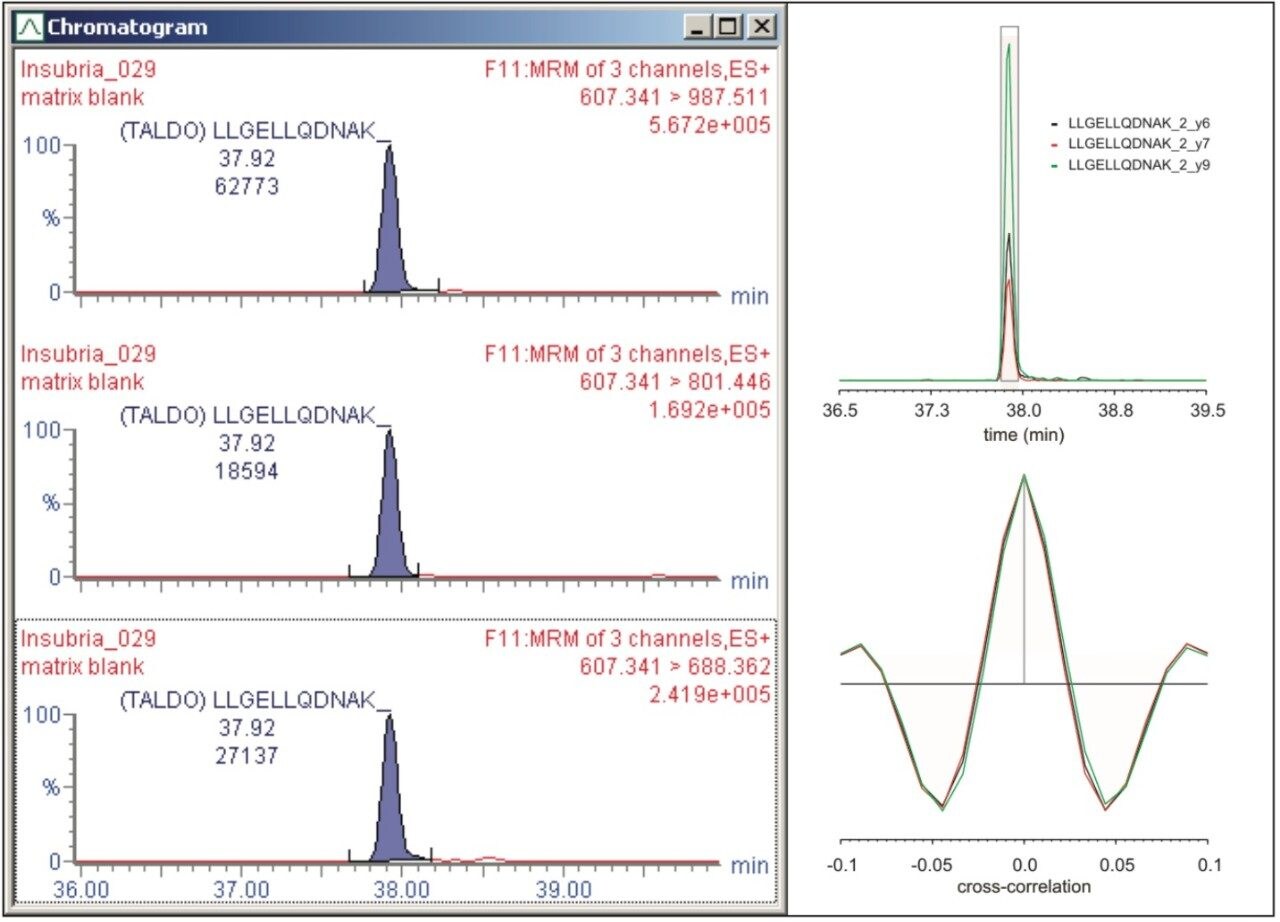
The resulting MRM chromatograms are shown in Figure 1, illustrating both good chromatographic performance and separation, as well as adequate MRM quantitation response for the majority of the proteins from the signature panel with a mere 100 ng of material loaded on-column. The relative intensities of the individual MRM transitions for one of the peptides of interest are illustrated in Figure 2. Throughout the complete experiment, comprising 147 LC-MS runs in total, including blanks and calibration standard runs, the relative areas of the individual transitions were constant, as shown by the inset of Figure 2 for LLGELLQDNAK. The average relative areas were 0.59 ± 0.01, 0.18 ± 0.02, and 0.23 ± 0.01 for y9, y7, and y6, respectively.