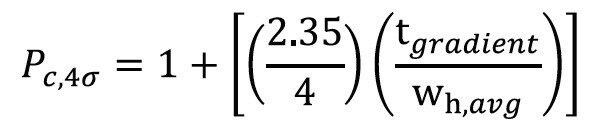Peptide Separations
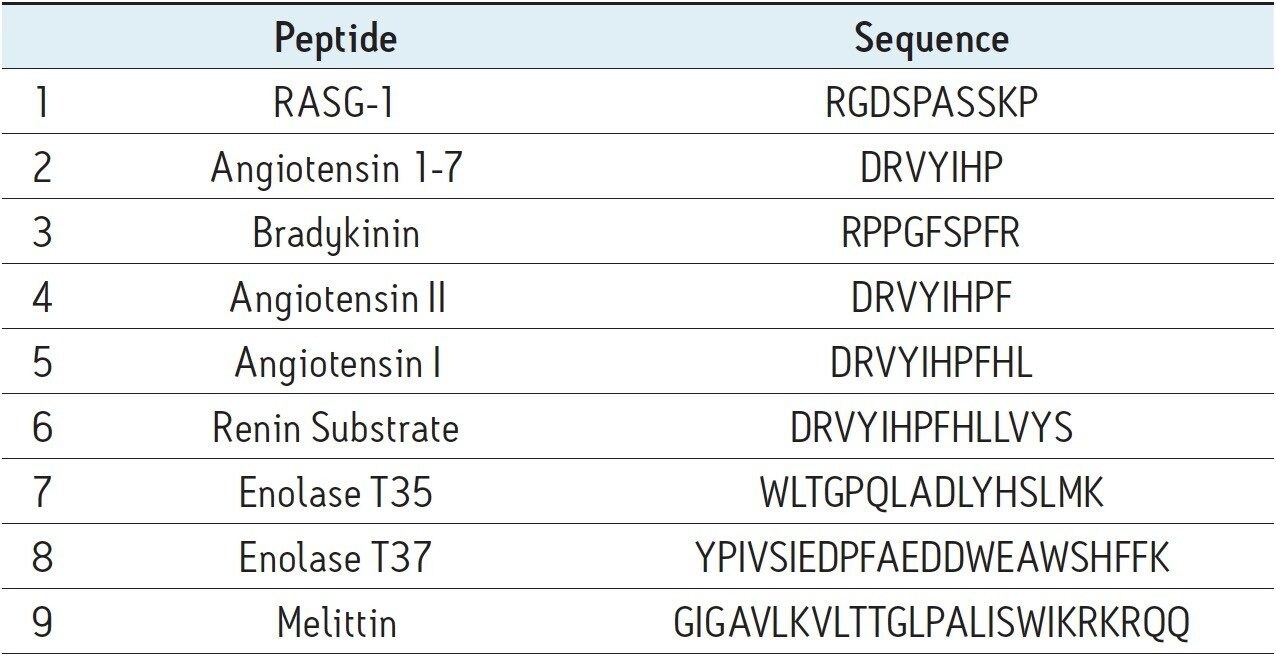
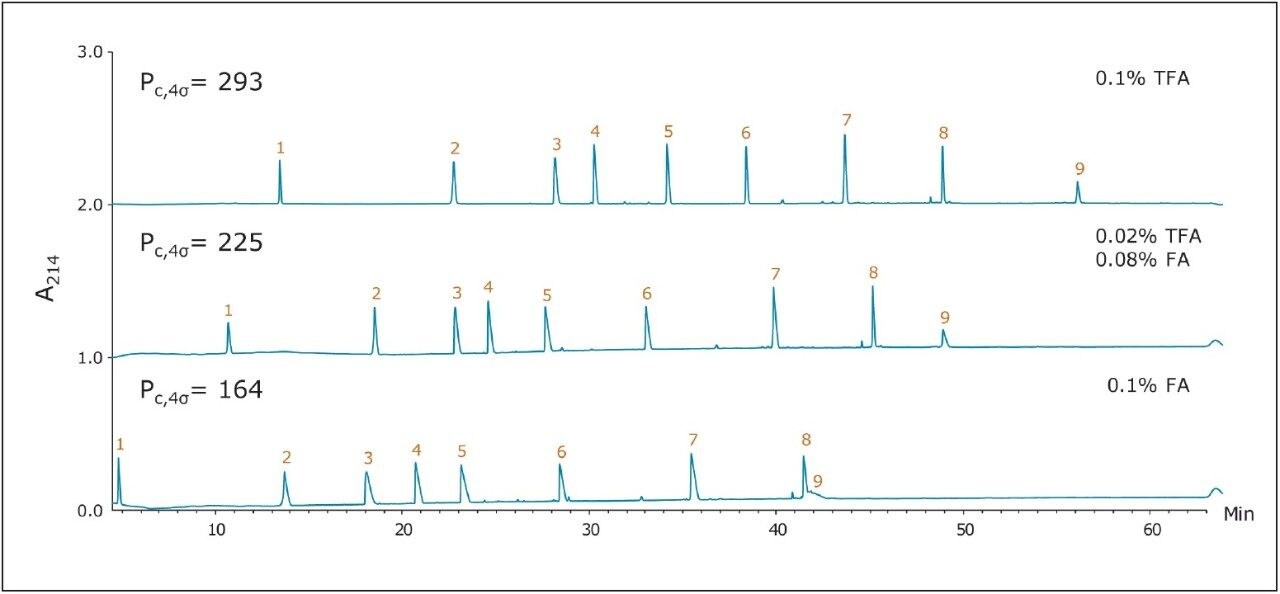
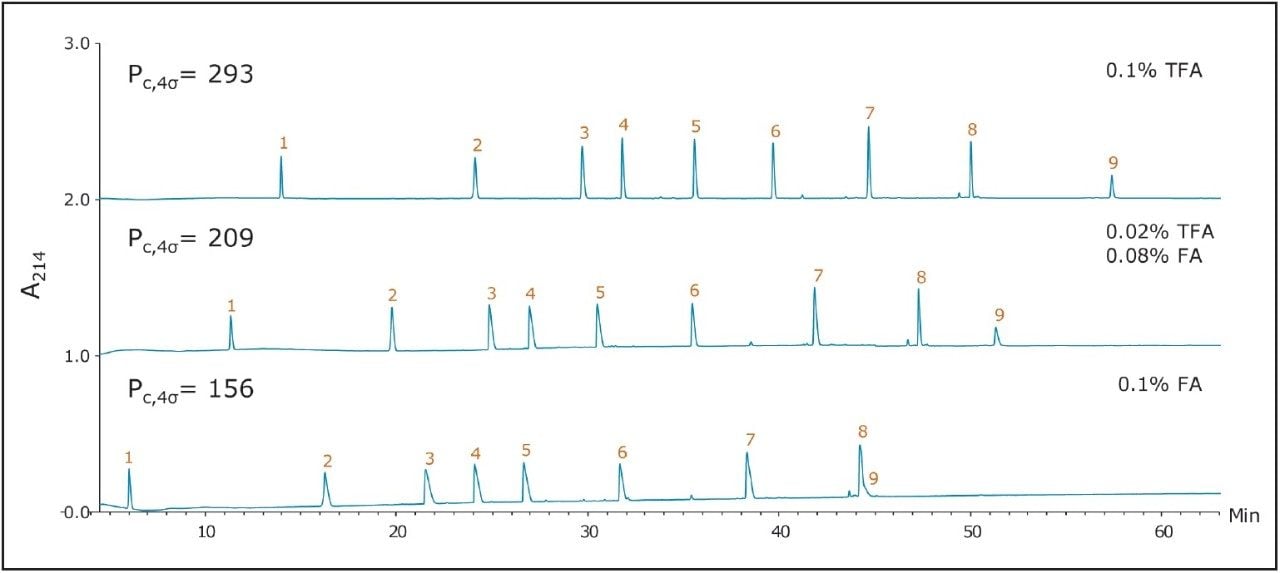
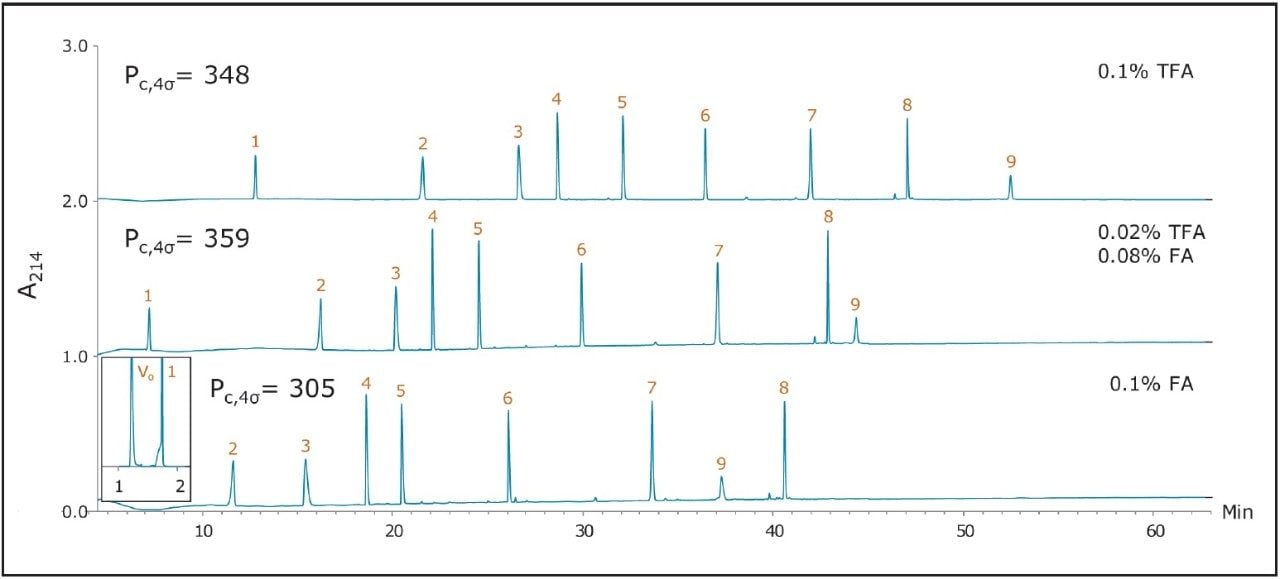
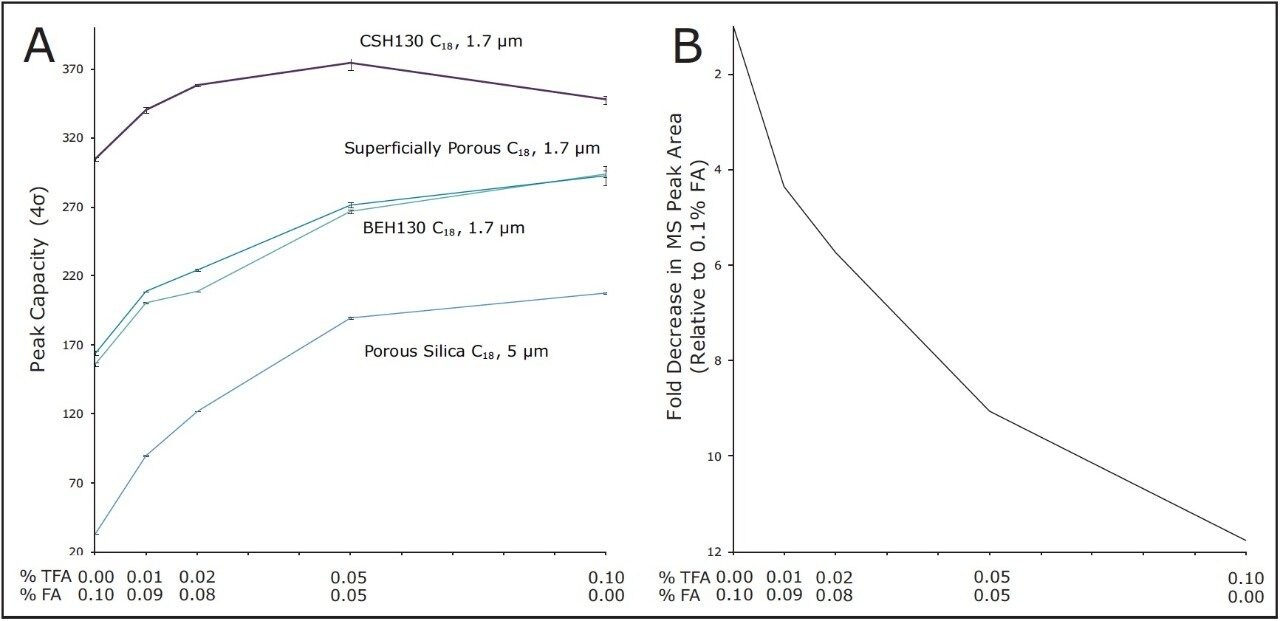
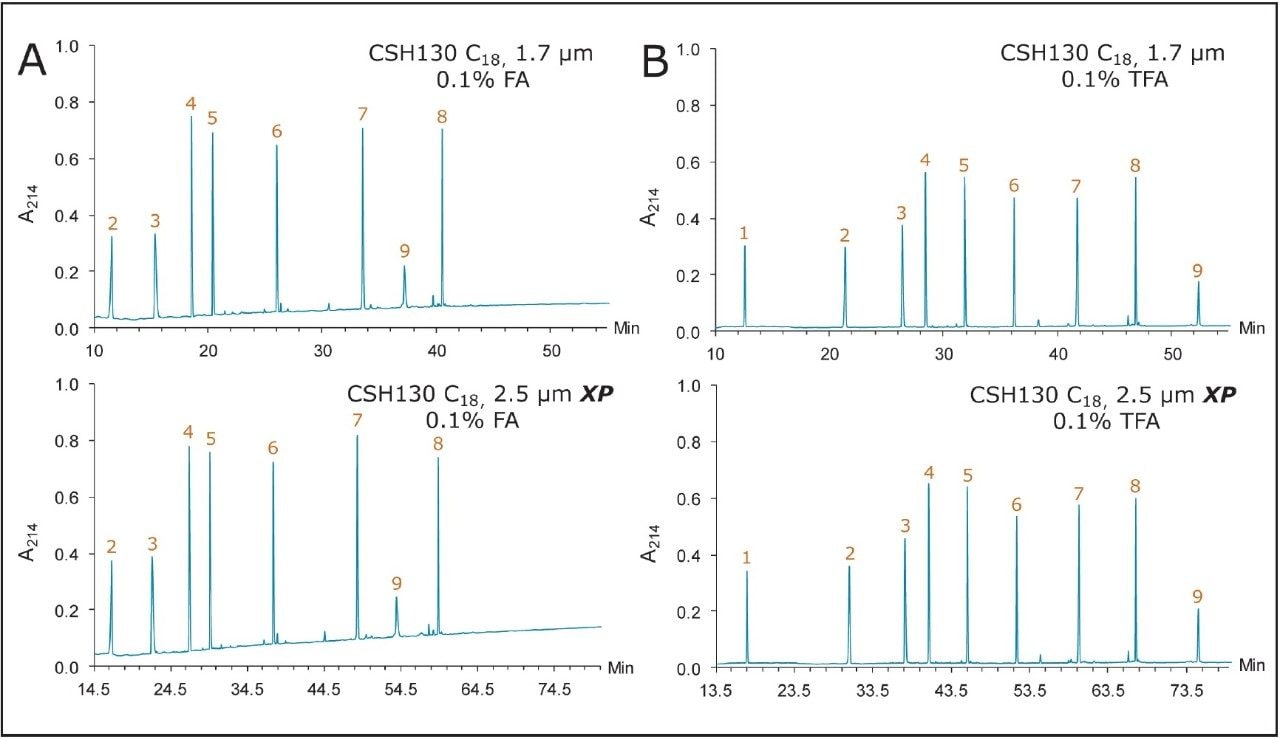
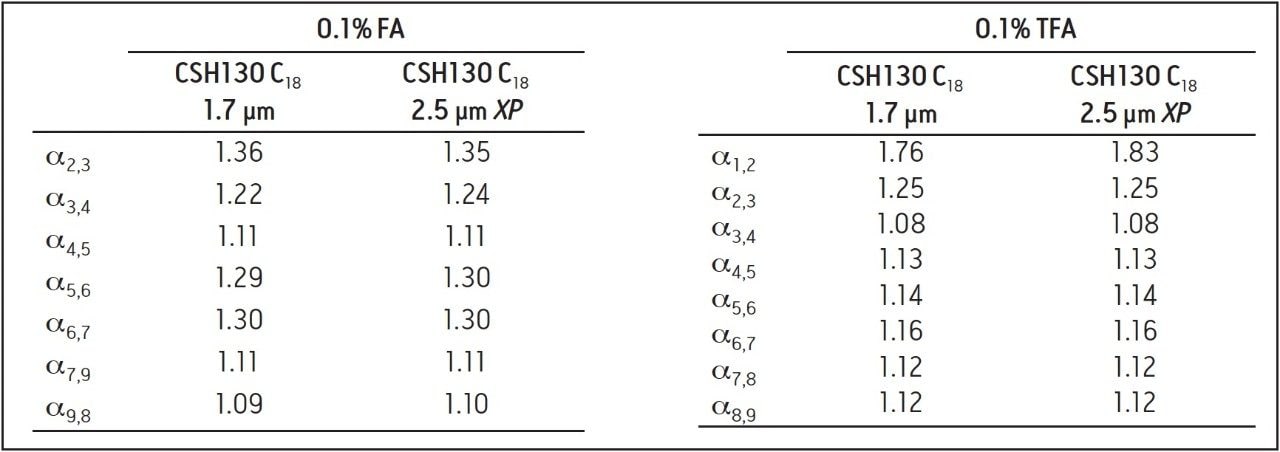
The MassPREP Peptide Mixture contains nine different peptides, varying in amino acid composition, mass, length, and polarity. Peptide sequences are shown in Table 1. Since this mixture is composed of such a diverse set of peptides, it is useful for evaluating mass spectrometric as well as chromatographic performance. To this end, the peptide mixture was employed to benchmark the separation performance of CSH130 C18 against other commonly used stationary phases for peptide analysis. Included in this study were phases with pore sizes large enough in diameter (100 to 300Å) to efficiently separate the peptides in the mixture, which are all less than 3 kDa in mass. In a separate application note, the use of CSH130 C18, with its 130Å pores for separating even larger polypeptides is discussed.11 For the sake of relevance, mass loads for the individual peptides in the mixture were made approximate to conditions common for peptide mapping of antibodies, wherein 20 to 50 μg of Lys-C or tryptic digests are typically analyzed on 2.0 or 2.1 mm I.D. columns.2,12-13 Similarly, separations were completed with mobile phases containing a strong ion pairing agent, TFA, a weaker ion pairing agent, FA, or a combination thereof. Performance under these various conditions was of interest, since RP peptide separations are routinely coupled with mass spectrometry. In such applications, formic acid is often preferred over TFA as a mobile phase additive because it permits more sensitive detection.14-15
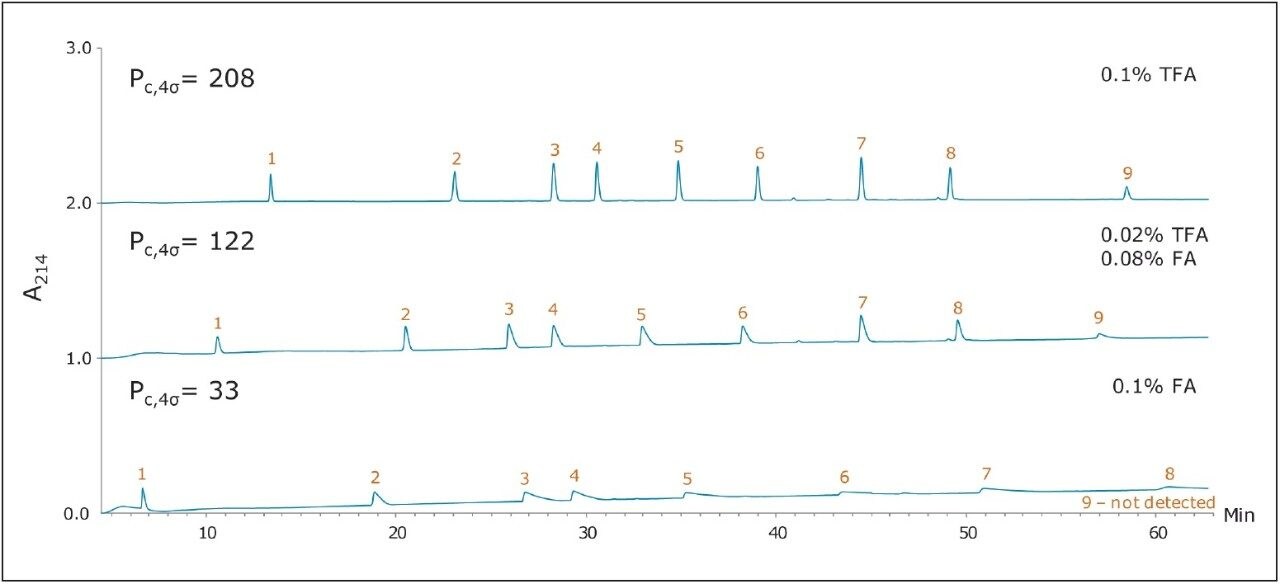
Figure 1 shows three chromatograms of the MassPREP Peptide Mixture obtained using a 5 μm silica C18 column under different mobile phase conditions. Separations with TFA provided peaks that were generally symmetrical. Most peaks did not exhibit excessive broadening or tailing. However, peak shape was found to be extremely poor with the FA containing mobile phases. In addition, the largest peptide in the mixture, melittin, was not detected, either because it eluted as too broad a peak or simply failed to elute. Results such as these are typical for HPLC-based peptide separations.