The safety of our food supply can no longer be taken for granted. As the world changes and populations continue to grow, so will the responsibility of organizations to meet the demand of safe food supplies.
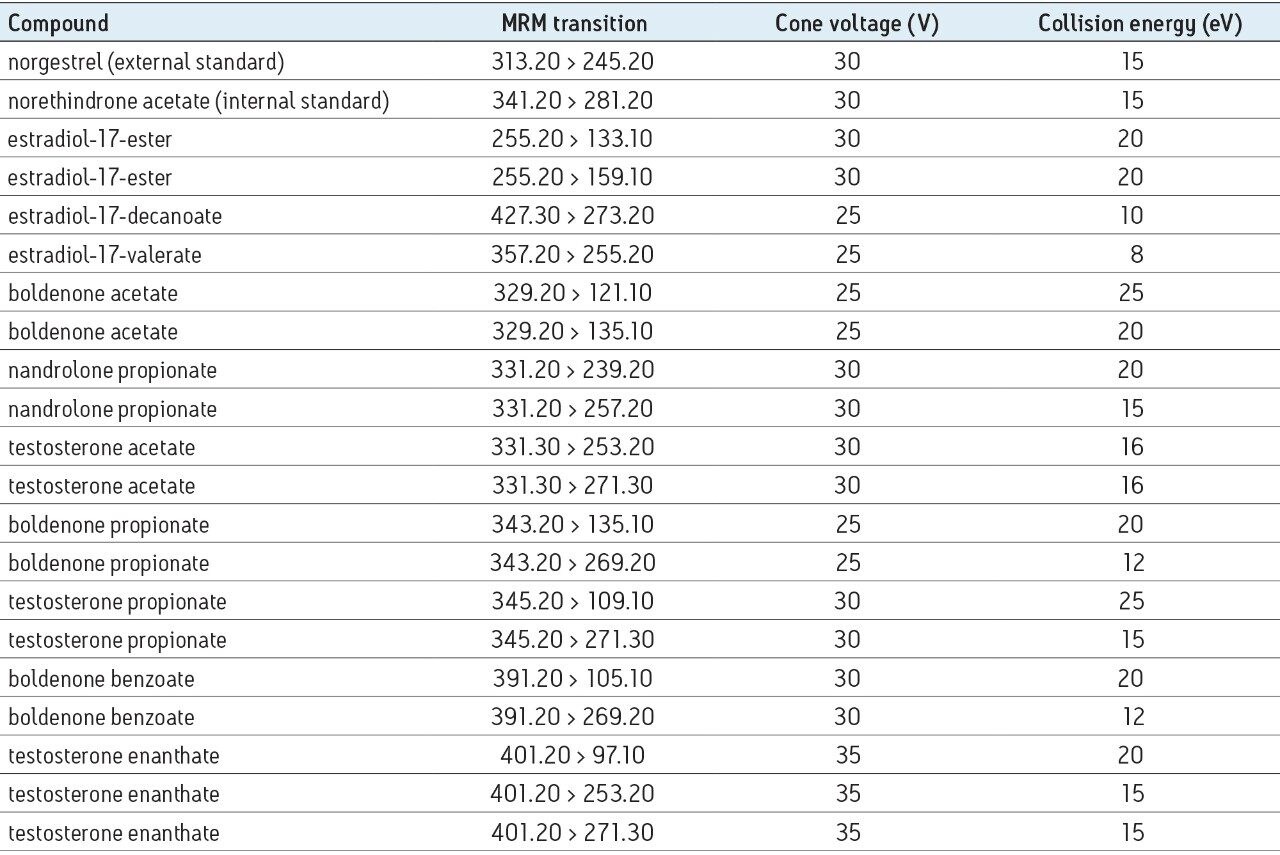
One route of human exposure to veterinary substances is through the food chain as a result of malpractice or illegal activities. The steroid esters are one group of substances causing concern, which might still be used as growth promoting agents, but are now forbidden from use in breeding animals in the European Union.1 No residues of these anabolic substances should be present in animal products.2
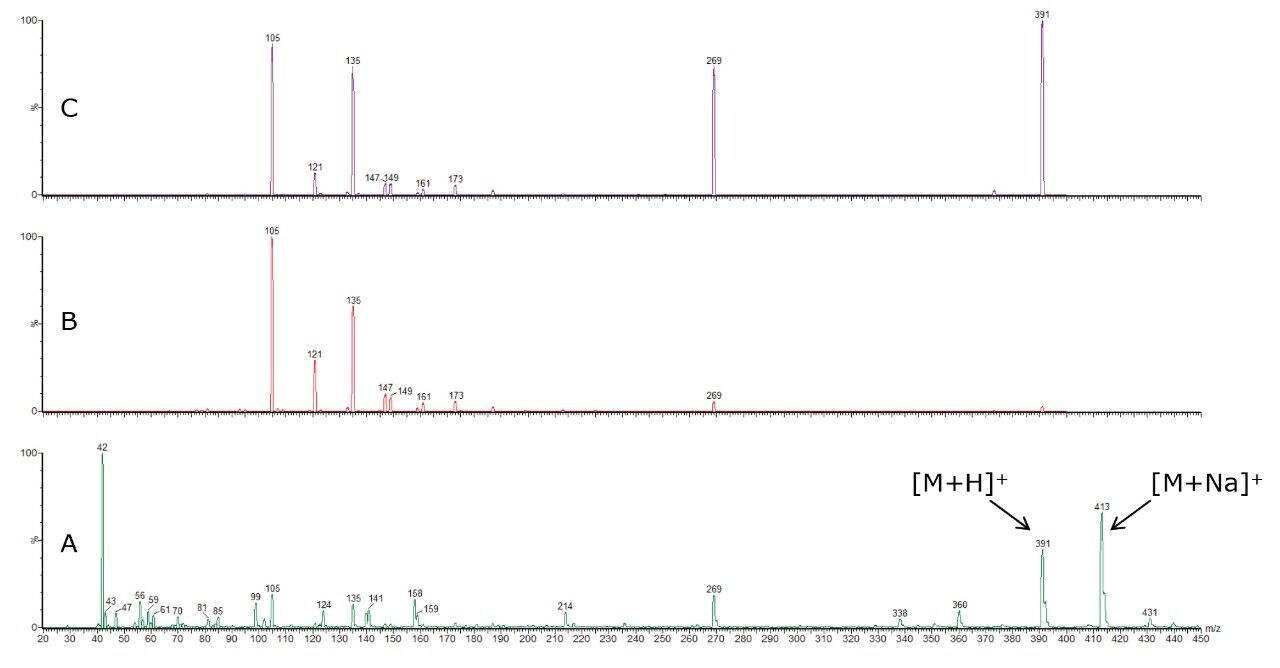
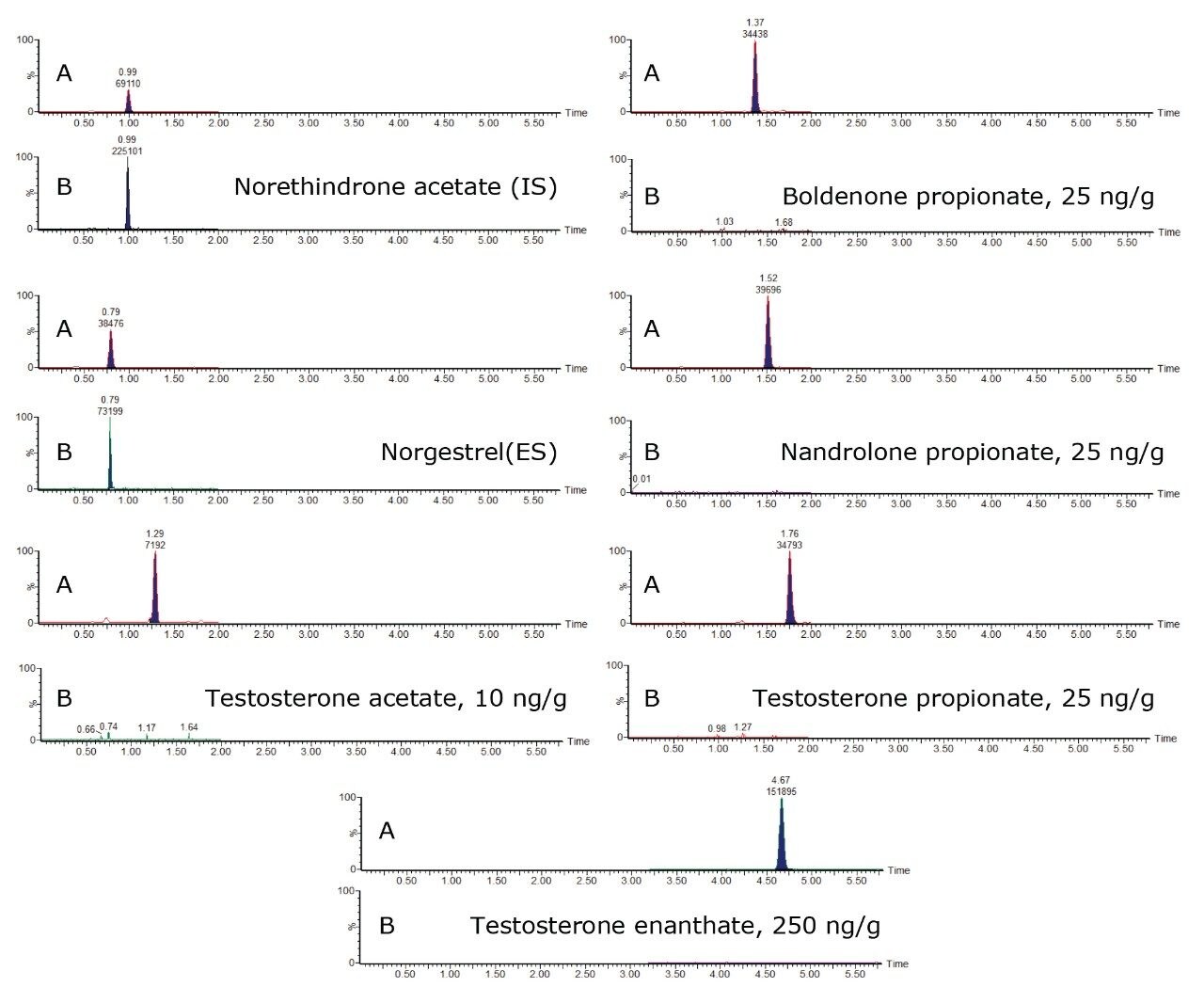
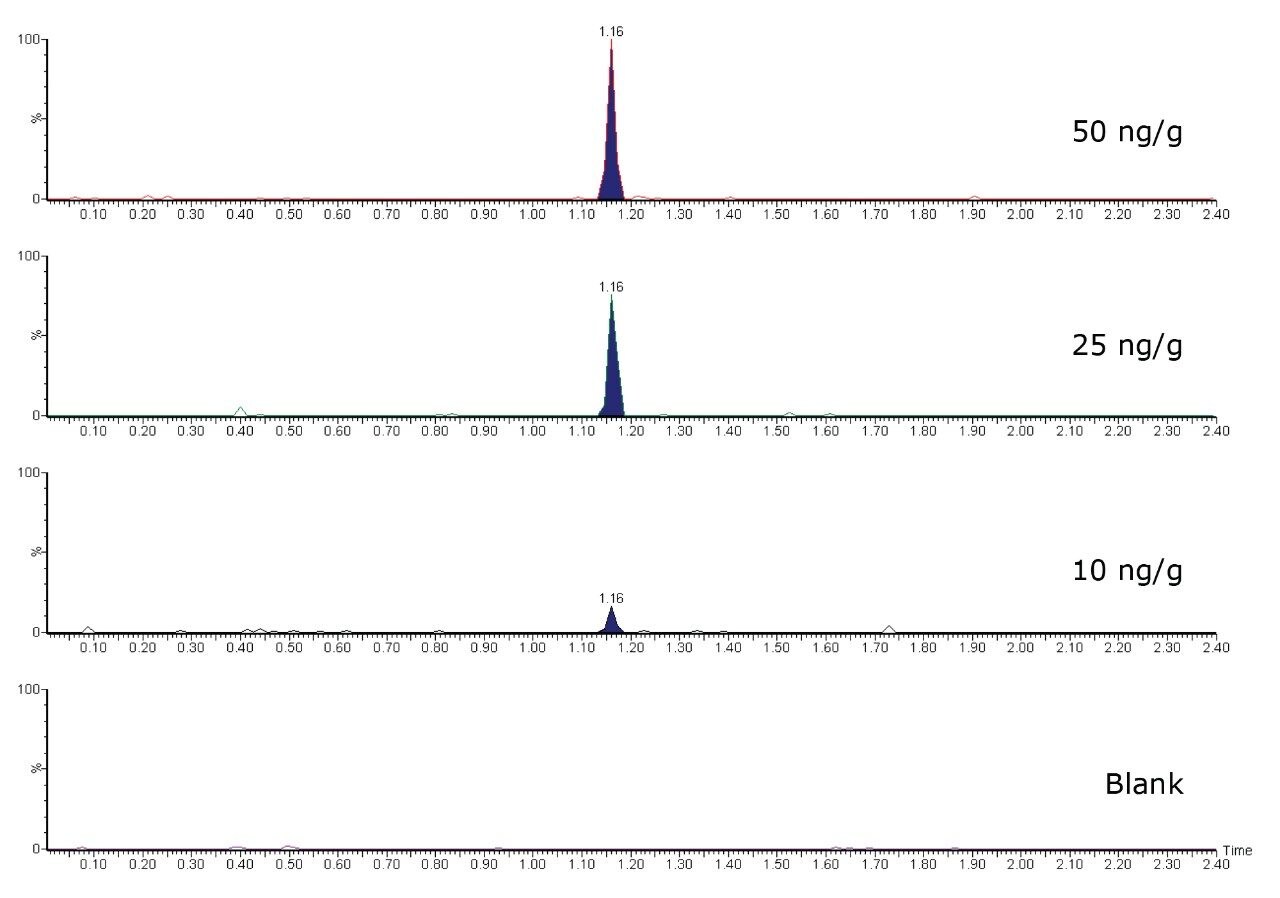
Many biological matrices, such as tissue or urine samples can be used for the control of steroid esters, but hair may be considered one of the most sensible matrices, due to its ease of collection and the ability to detect residues for a long time after administration of some substances.3-6 In urine, it is hard to differentiate between the metabolites of ever-present endogenous natural steroids, and the identical metabolites of natural steroids from the hydrolyzed esters.7 Furthermore, natural steroids are usually administered as synthetic steroid esters that are rapidly hydrolyzed in vivo into natural steroids. Nevertheless, the identification of the synthetic ester form in hair provides an interesting, efficient, and additional confirmatory method to overcome any inconclusive urine analyses. Indeed, the ability to detect and identify the compound itself for a long time after administration is a strong argument in delivering a confident analytical result. The main problem with this approach is that the concentrations of the steroid esters are extremely low (low ppb range); and the sample size limited (for practical reasons of sample collection, e.g. 100 mg hair).
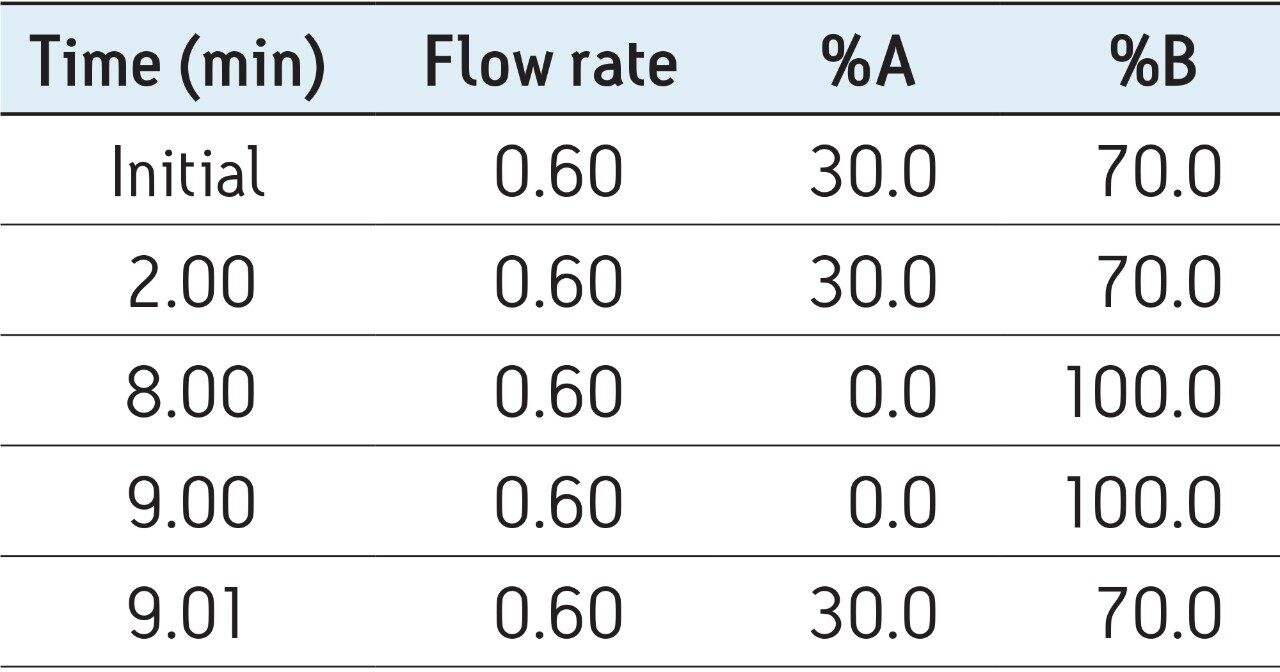
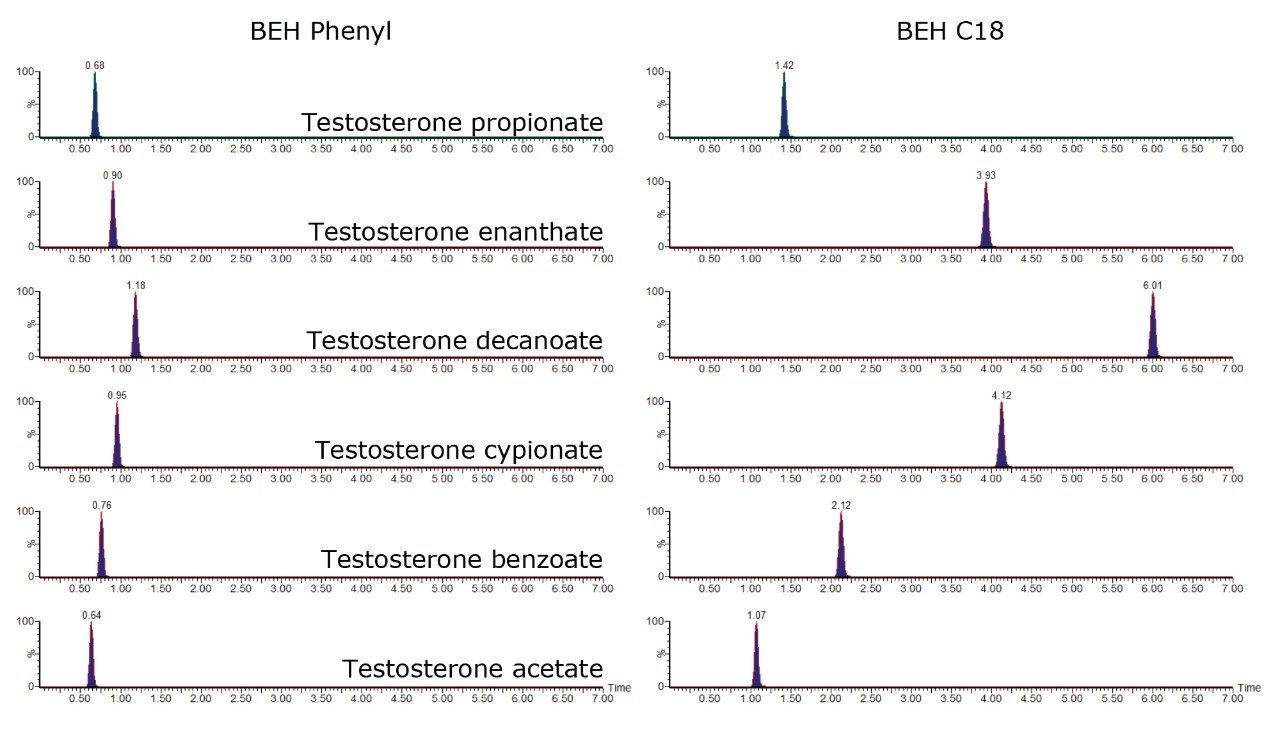
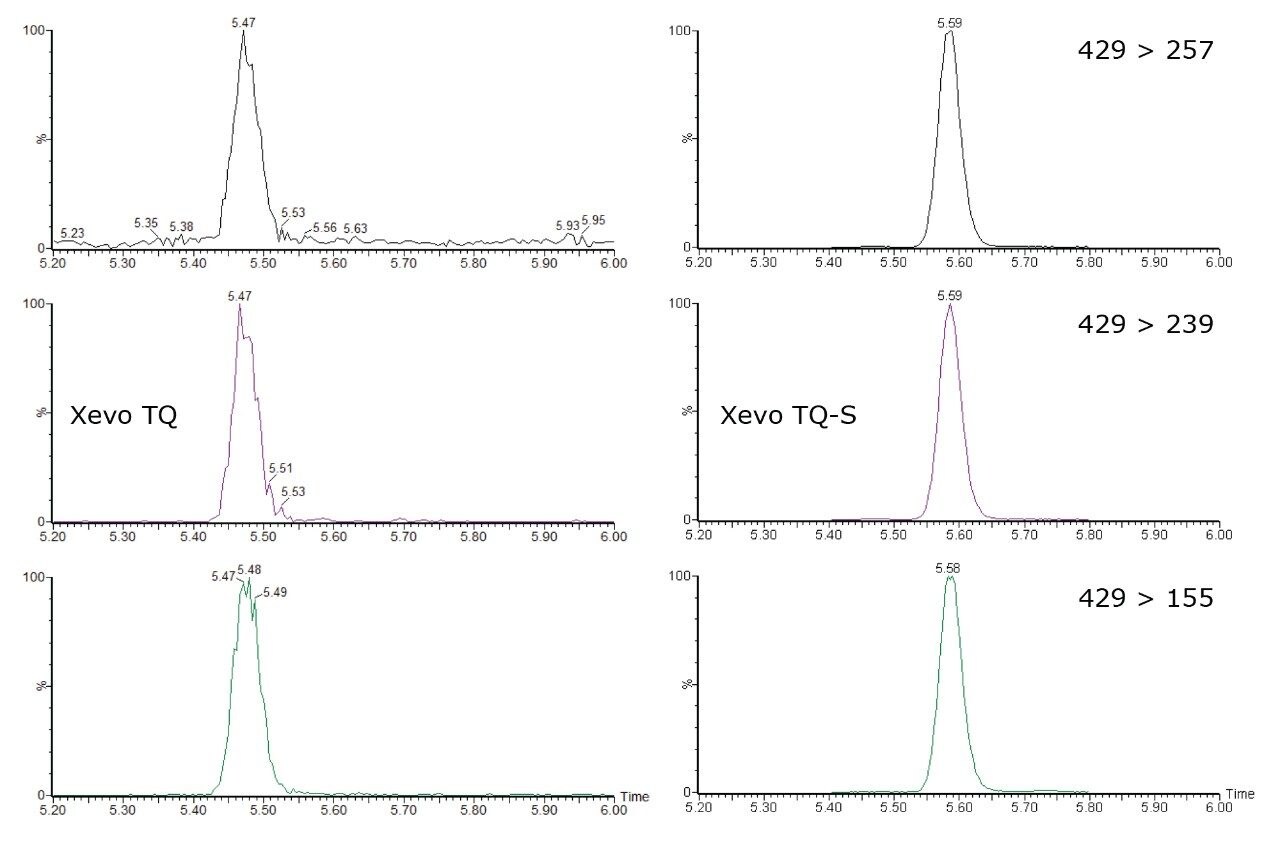
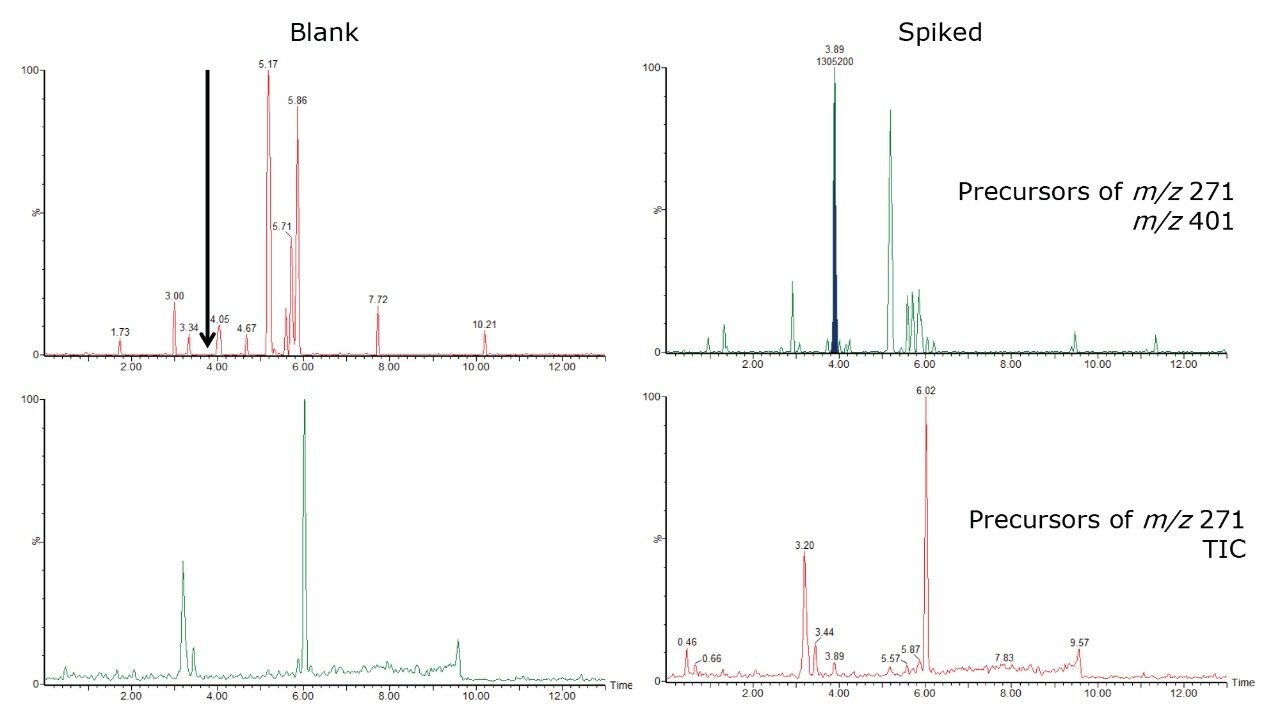
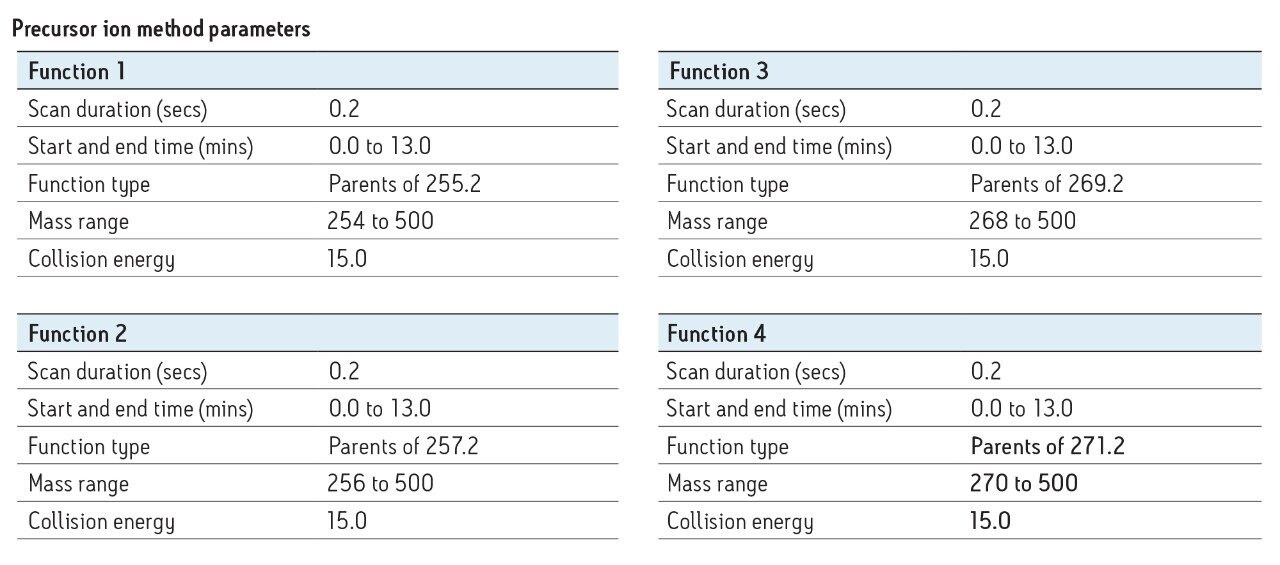
In the literature, either GC-MS/MS5 or LC-MS/MS7-9 could provide the detection of steroids esters to relevant levels. The less volatile compounds, such as benzoate or decanoate steroids, do not give expected results in GC-MS/MS in routine analysis, because the instrument has to be under very clean and optimized conditions to reach the validation performance.5 Furthermore, the analysis resulted in a 40 minute experiment.5 Given these reasons and the wish for an additional analytical technique for confirmation, we investigated the analysis of steroid esters in cattle hair by LC-MS/MS.