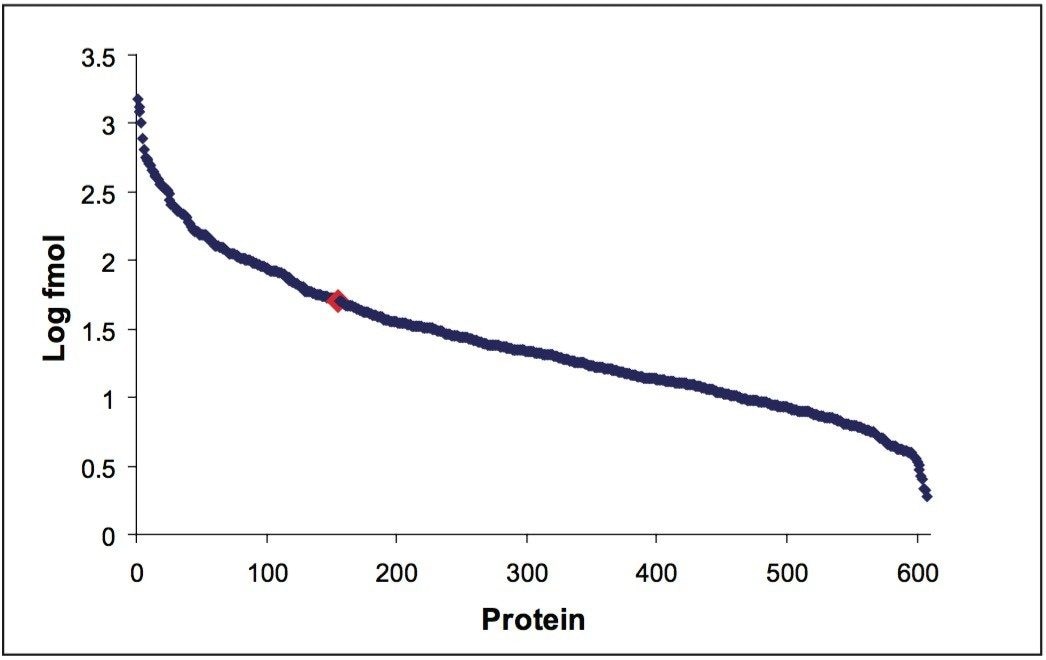
A biomarker is measured as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic intervention. Many times, a putative biomarker is a protein or peptide that is expressed at a relatively low level compared to the surrounding proteome. The constitutive or housekeeping proteins are present in concentrations that are orders of magnitude above the protein of interest, which makes identification and quantitation difficult. In order to validate a candidate biomarker, many samples need to be analyzed to prove that the same analytes are reproducibly identified and are changing in a statistically significant manner due to a perturbation.
Two-dimensional (2D) chromatography is often used to separate peptides from proteomic samples in a biomarker discovery workflow. 2D chromatography of tryptic peptides has traditionally been performed using strong cation exchange (SCX) followed by reversed-phase (RP) chromatography, due to the orthogonal nature of the separation mechanisms of these two techniques. The major disadvantage of this approach is that peptides are often split across first dimension SCX fractions due to the relatively low resolution of peptides on SCX material.
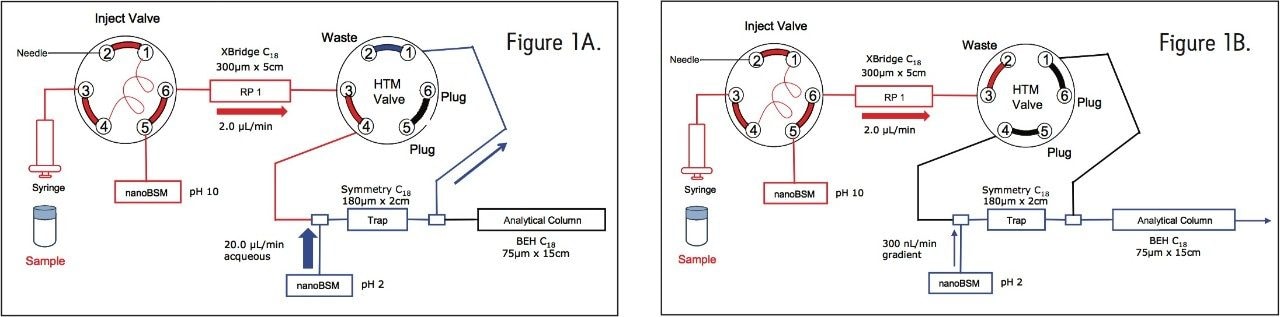
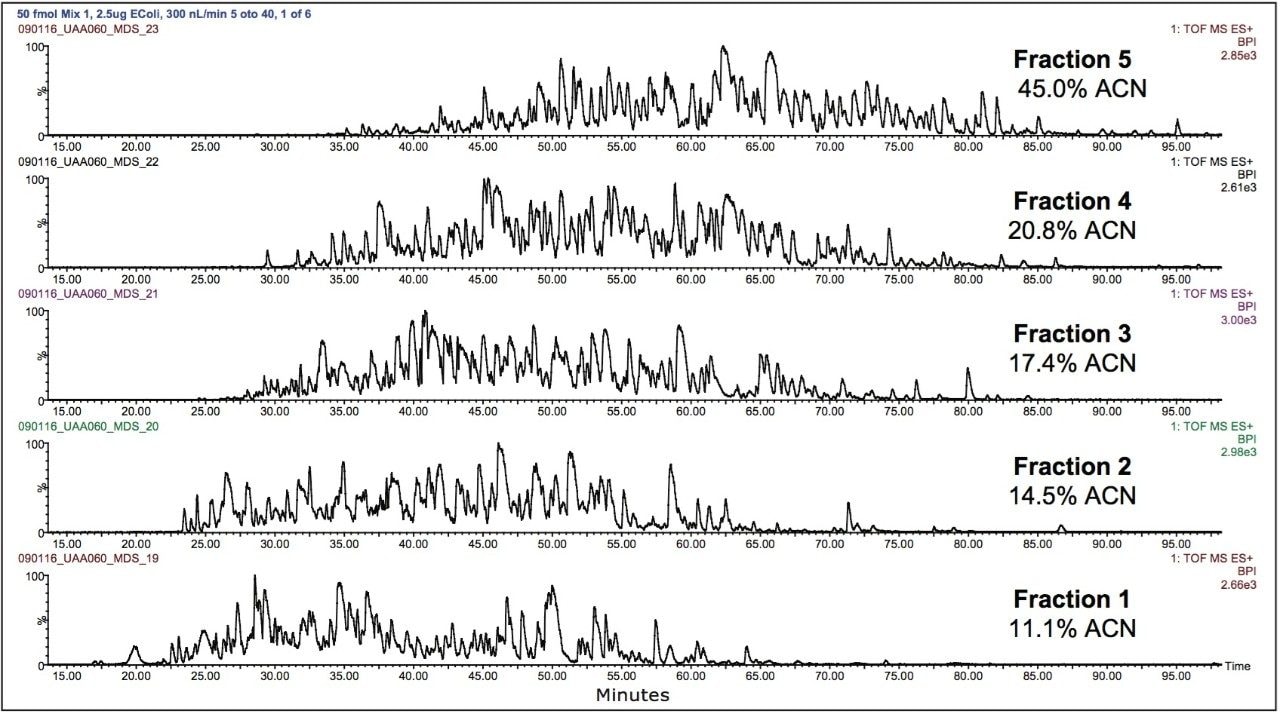
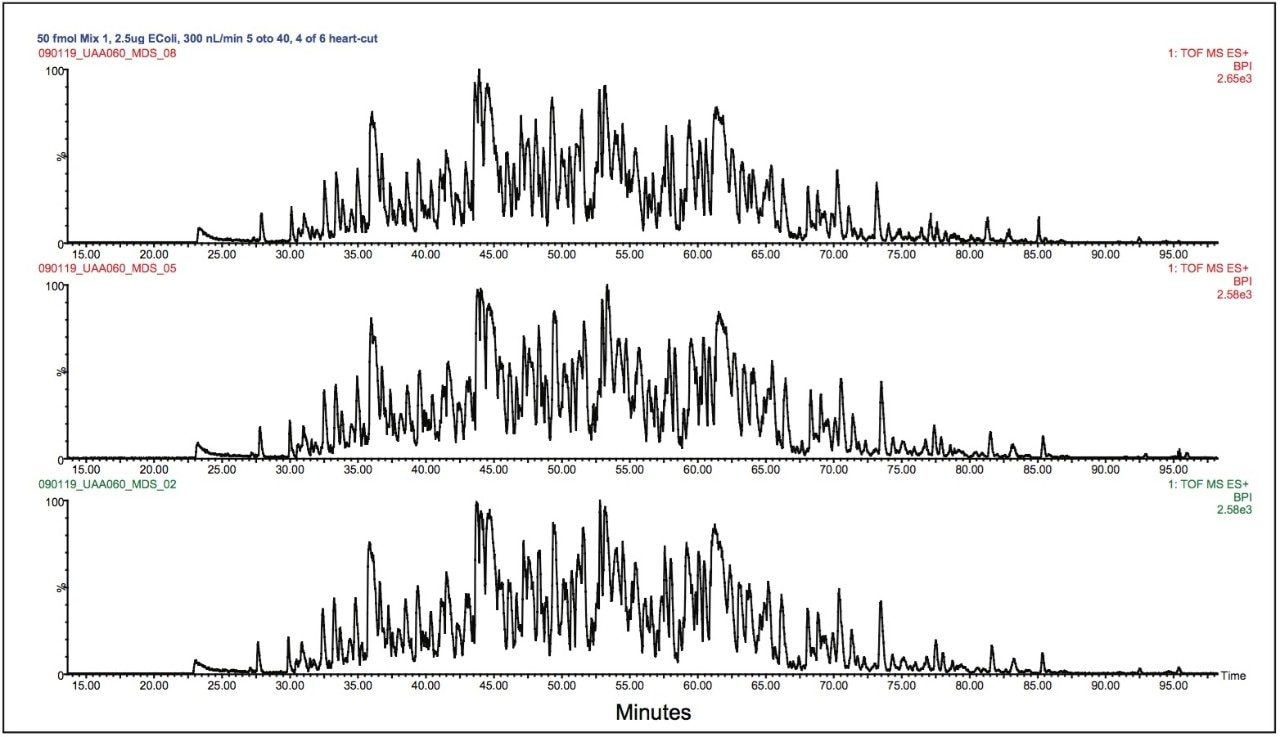
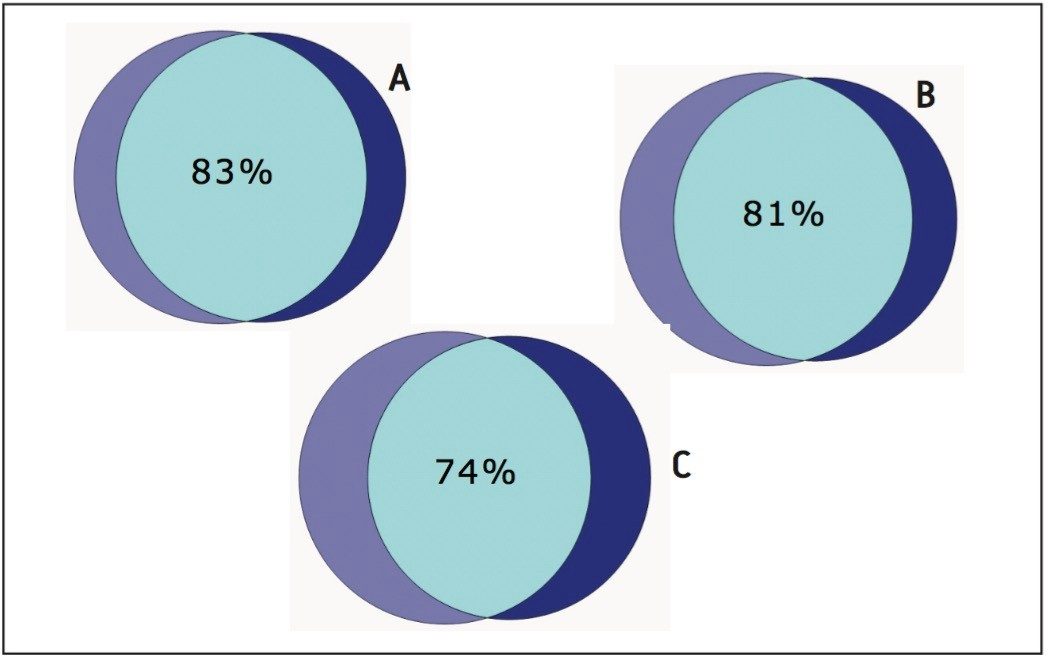
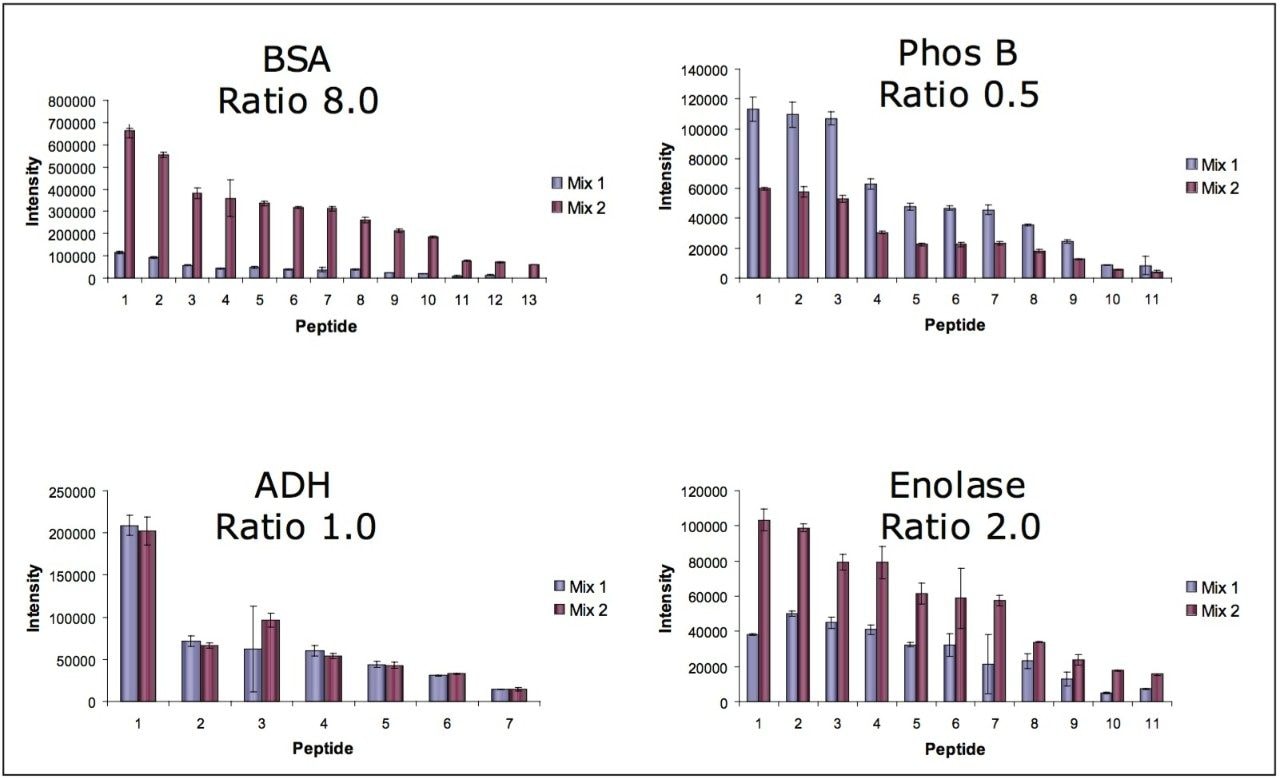
A highly reproducible method for performing online 2D chromatography with mass spectrometry has been developed where peptides are separated by RP chromatography at high pH in the first dimension, followed by an orthogonal separation at low pH in the second dimension. An online dilution of the effluent was performed after the first dimension to ensure no peptides were lost during trapping prior to the second dimension. For targeted biomarker validation, running an entire 2D experiment would be time consuming given the limited number of target molecules that might need to be monitored and the number samples in a typical validation experiment. A preferred approach is to elute the targeted peptides in one fraction in a heart-cut manner. This application note will illustrate the application of online high/low pH RP/RP chromatography for heart-cut analysis.