Veterinary drugs are used in animal husbandry and aquaculture for therapeutic or disease-preventive reasons and, in some cases, to promote growth of livestock. However, when specified withdrawal periods are not observed, unsafe antibiotic residues, or their metabolites, may be present in edible products such as milk, eggs, and meat. To meet growing demand, shrimps and other seafood are often cultivated by aquafarms, where many animals are kept in relatively small spaces, making them more prone to diseases. In order to preserve animal health as well as to ensure production and to increase yields, antibiotics are used on a large scale. Residues of these antibiotics in foods of animal origin are a major concern because they are harmful to the consumer’s health and could induce pathogens to develop resistance.1
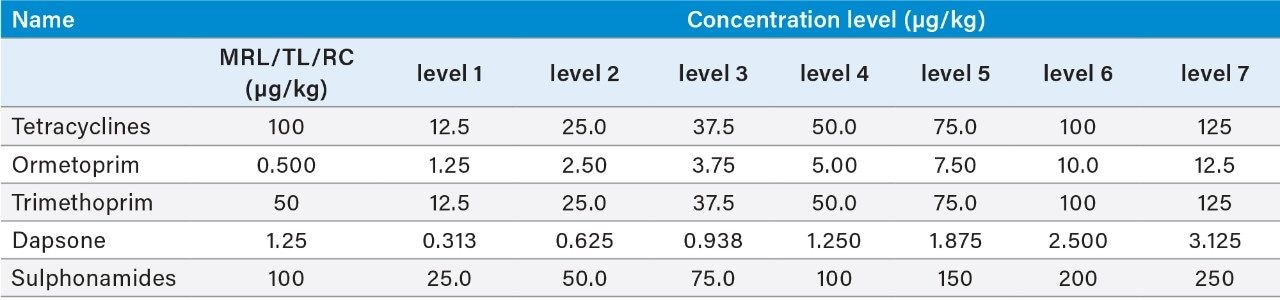
Authorities regulate the use of veterinary drugs by setting the maximum residue limits (MRLs) or by prohibiting the use of many substances to ensure the safety of the food and facilitate international trade between countries. In the EU, there are MRLs for a number of substances, which apply to all food-producing species; sulfonamides, expressed as the combined total residues of all substances within the sulfonamide group, and for tetracyclines, which relate to the sum of parent drug and its 4-epimer except for doxycycline (DC).2 Dapsone, demeclocycline, and ormetoprim are not approved for use on food-producing animals in the EU, and as such, have no MRLs, although dapsone has a recommended concentration (RC) of 5 μg/kg.3 When the substance is prohibited, the CCα and CCβ limits should always be as low as reasonably achievable (ALARA).
Surveillance combined with the effective enforcement, investigation, and inspection activities ensures the safe and effective use of veterinary medicines in various parts of the world. For example, EU countries must implement residue-monitoring plans to detect the illegal use or misuse of authorized veterinary medicines in food producing animals. These countries must also investigate the reasons for residue violations. Non-EU countries exporting into the EU must also implement a residue monitoring plan that guarantees an equivalent level of food safety.
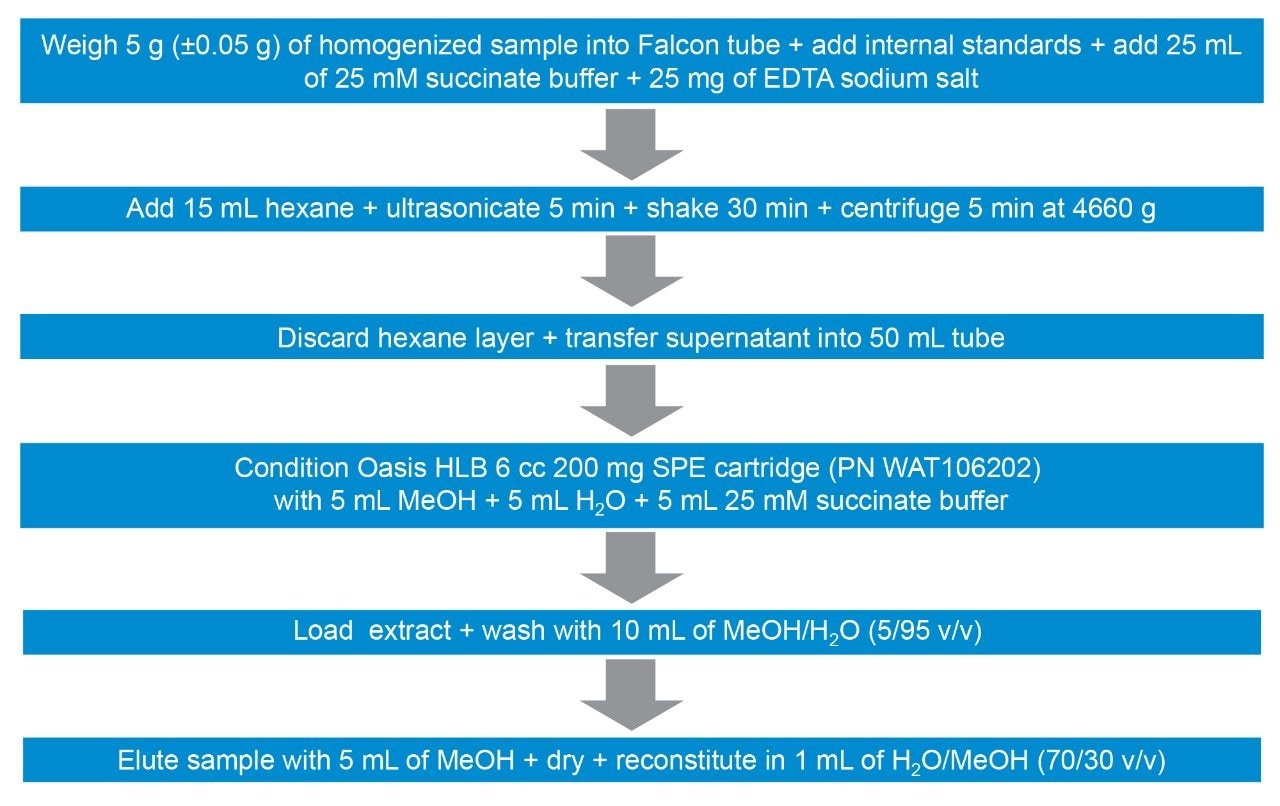
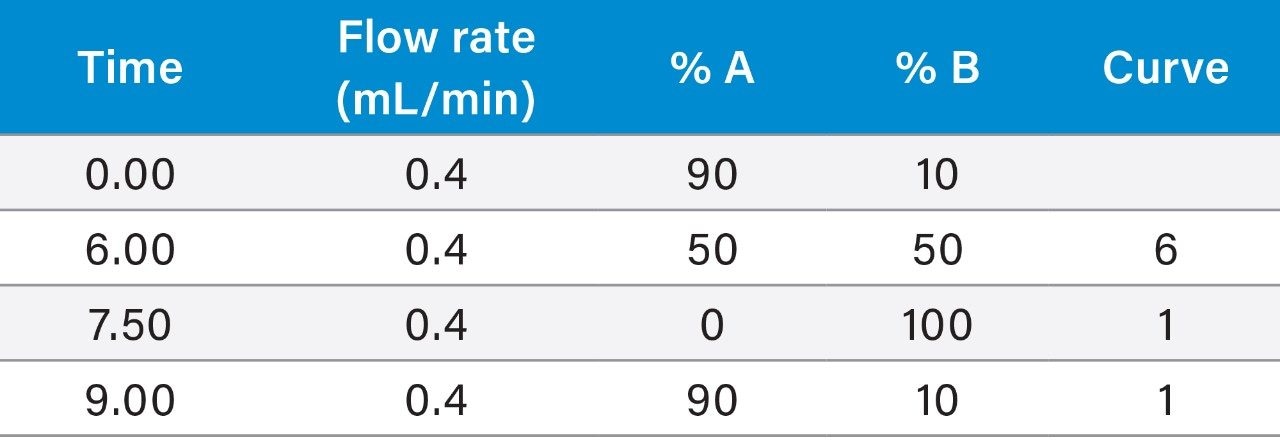
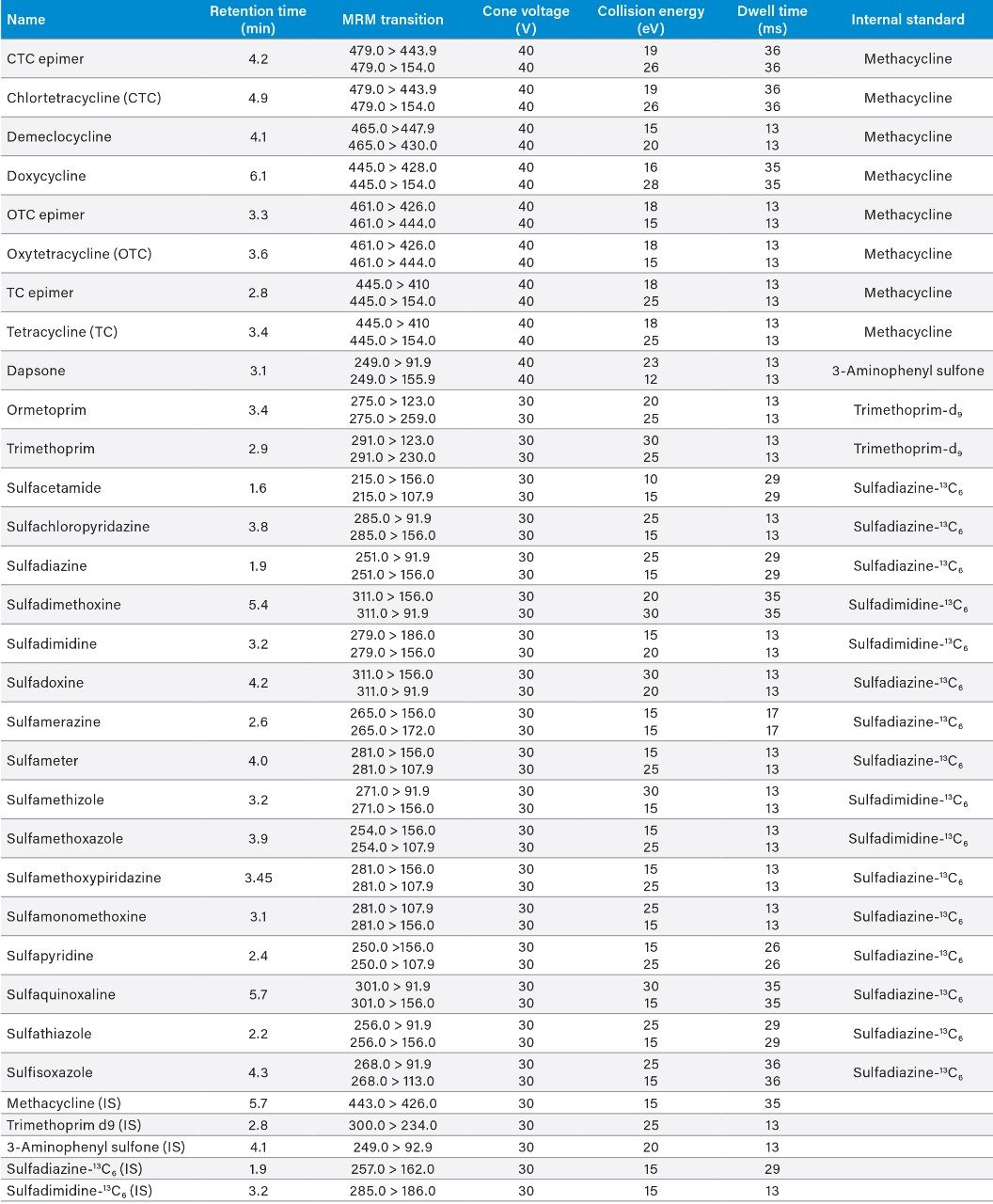
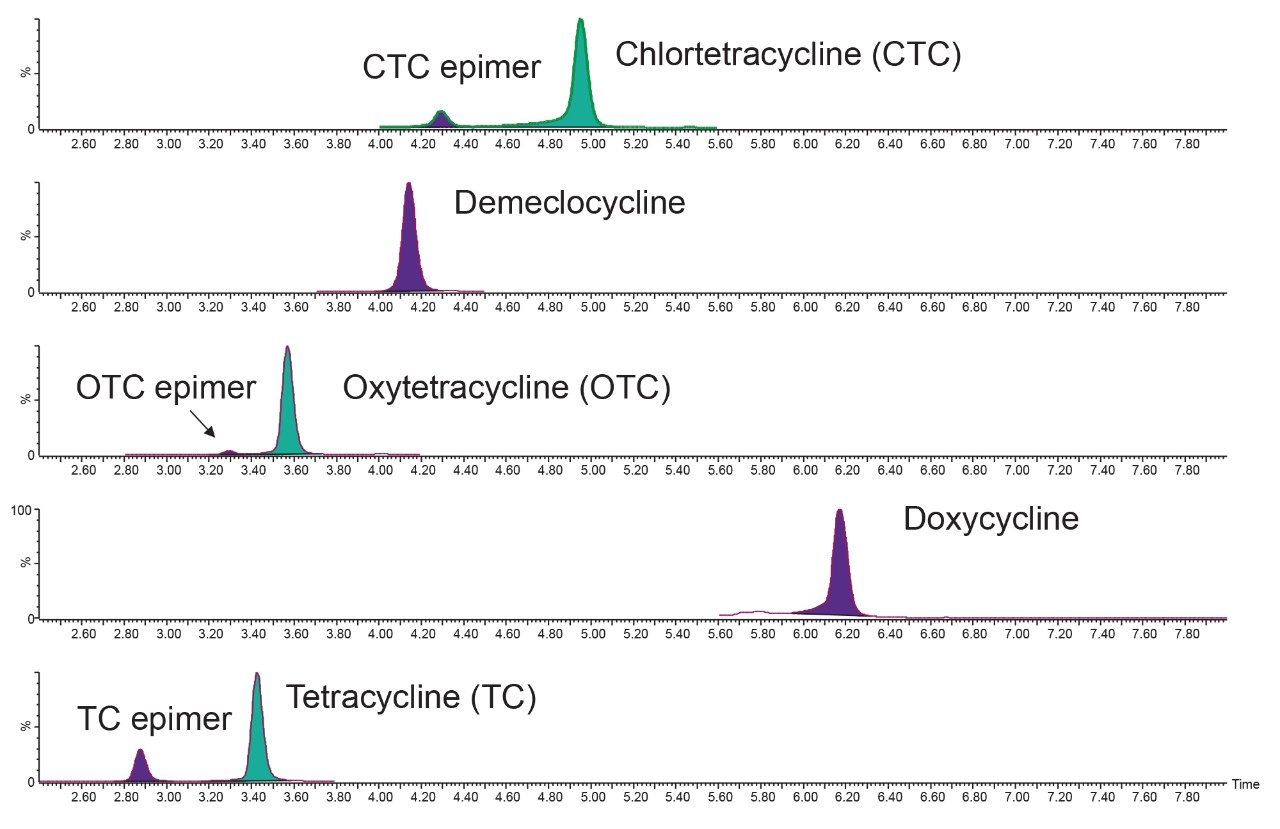
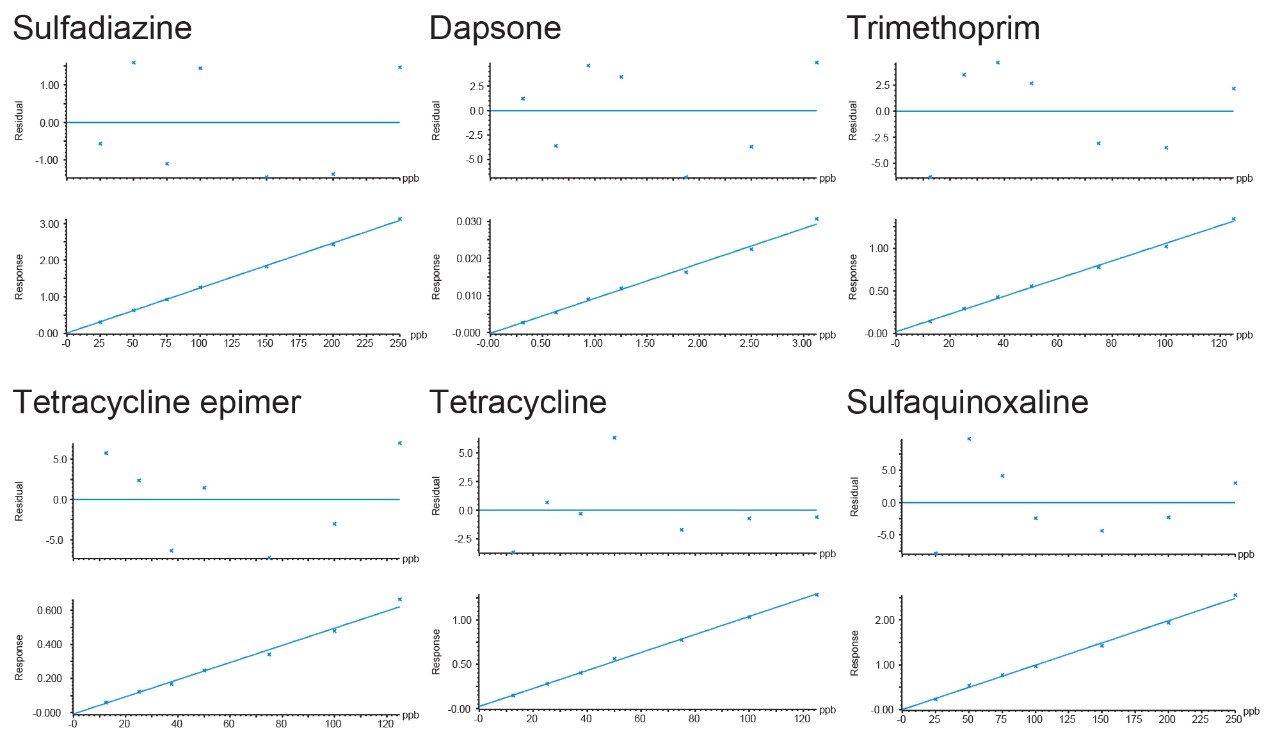
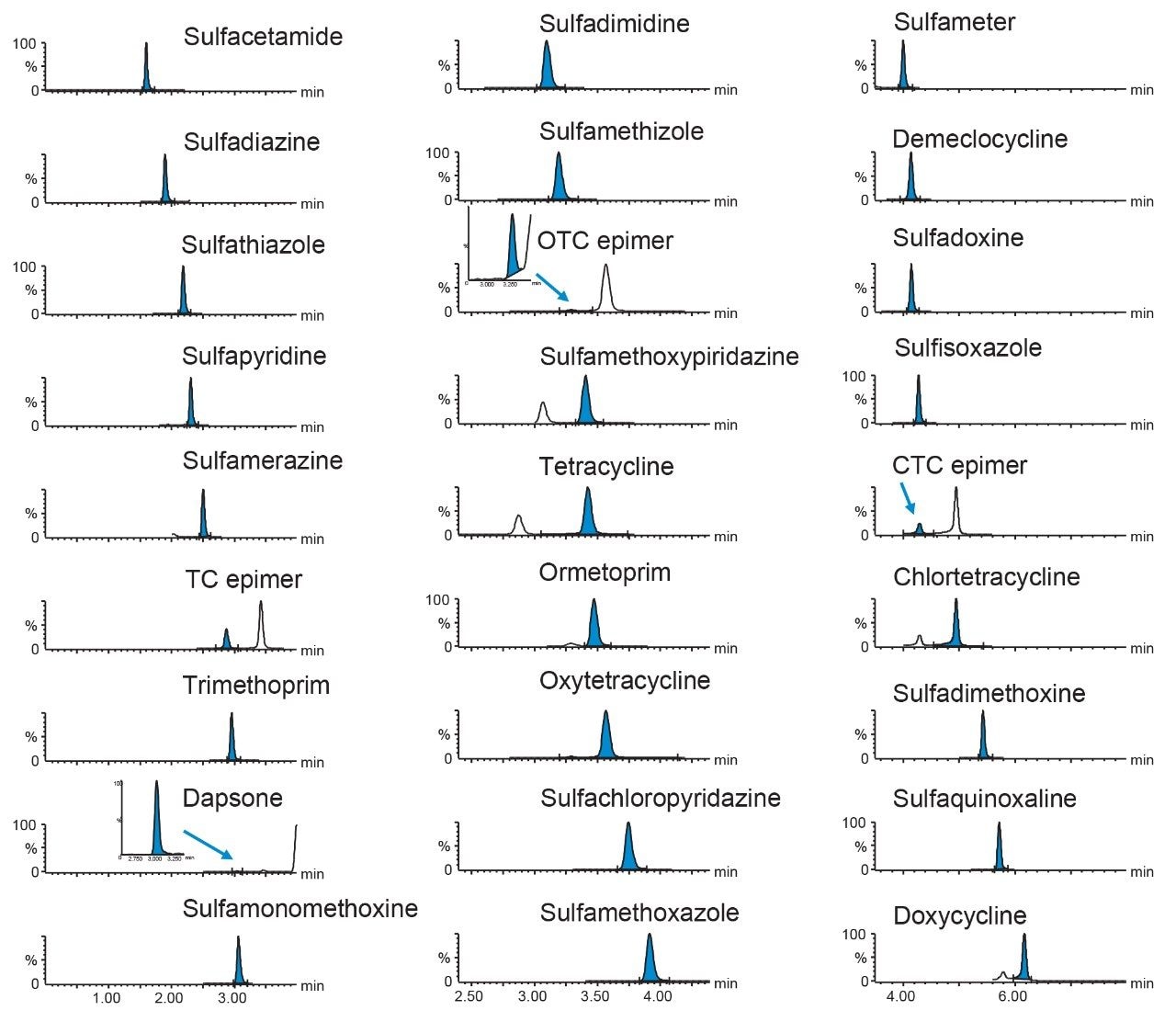
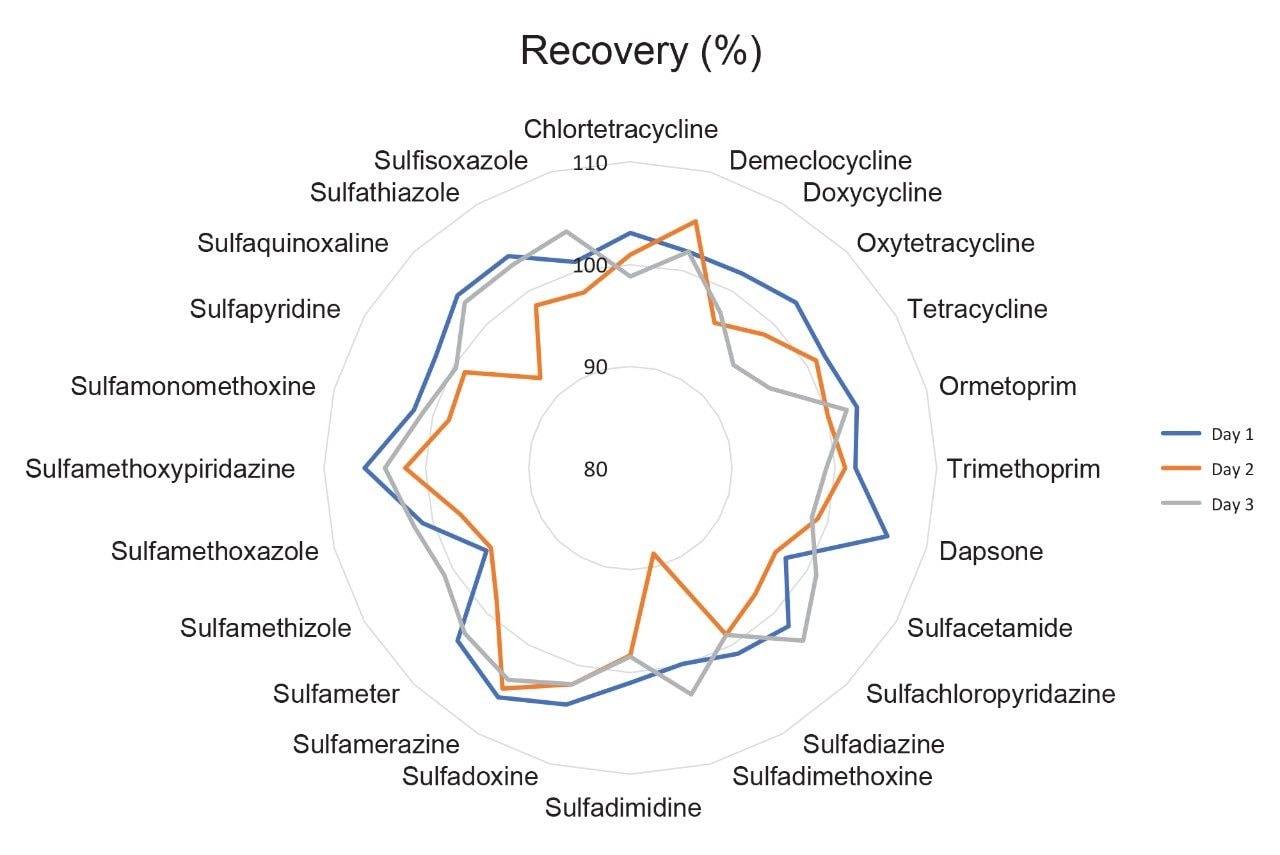
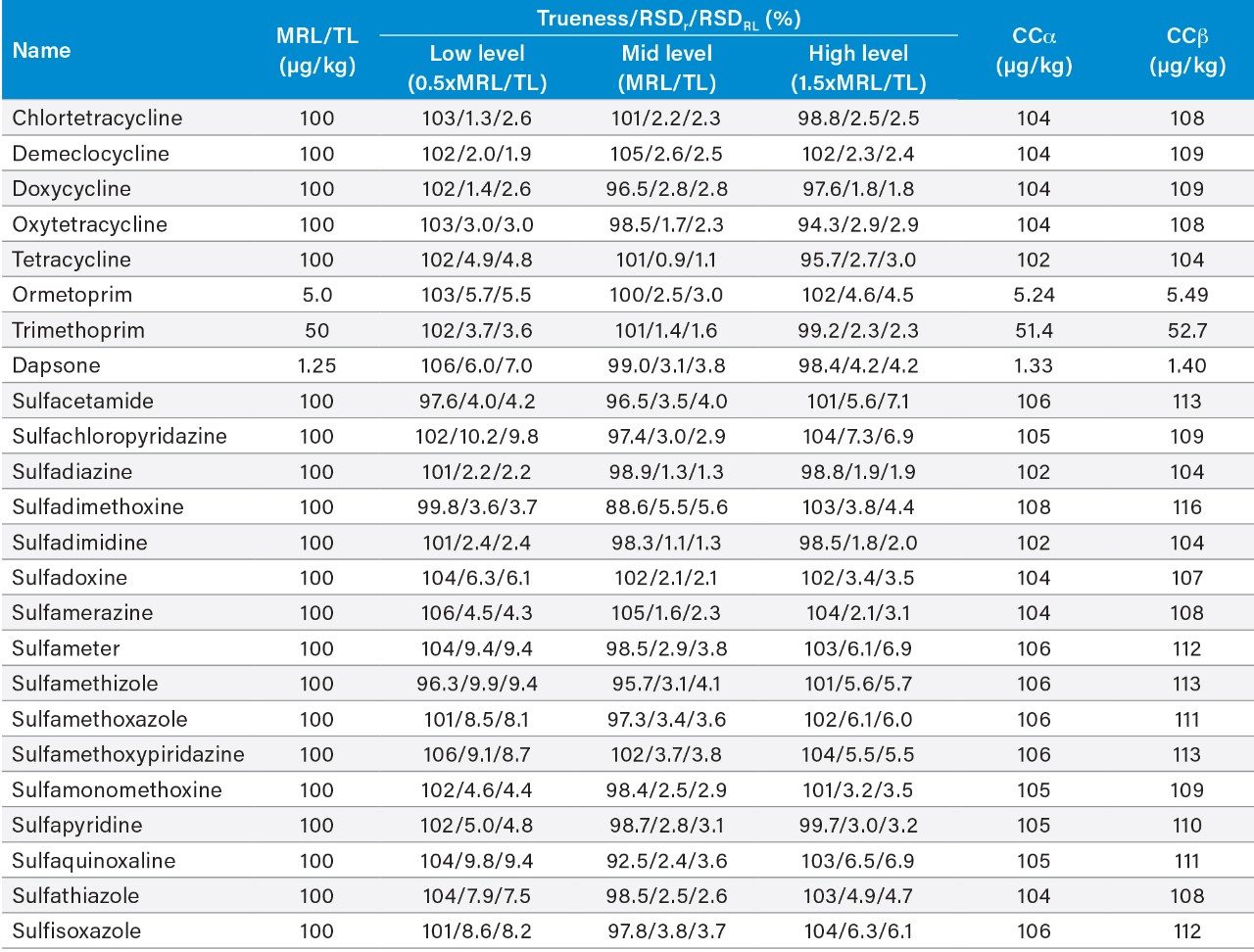
It is important, therefore, to develop a simple but accurate method for the determination of residues of a range of antibiotics in seafood. This application note describes the results of a successful validation of the analysis of shrimp tissue for tetracyclines, sulfonamides, trimethoprim, ormetoprim, and dapsone using the Waters ACQUITY UPLC I-Class PLUS System coupled to the Xevo TQ-S micro.