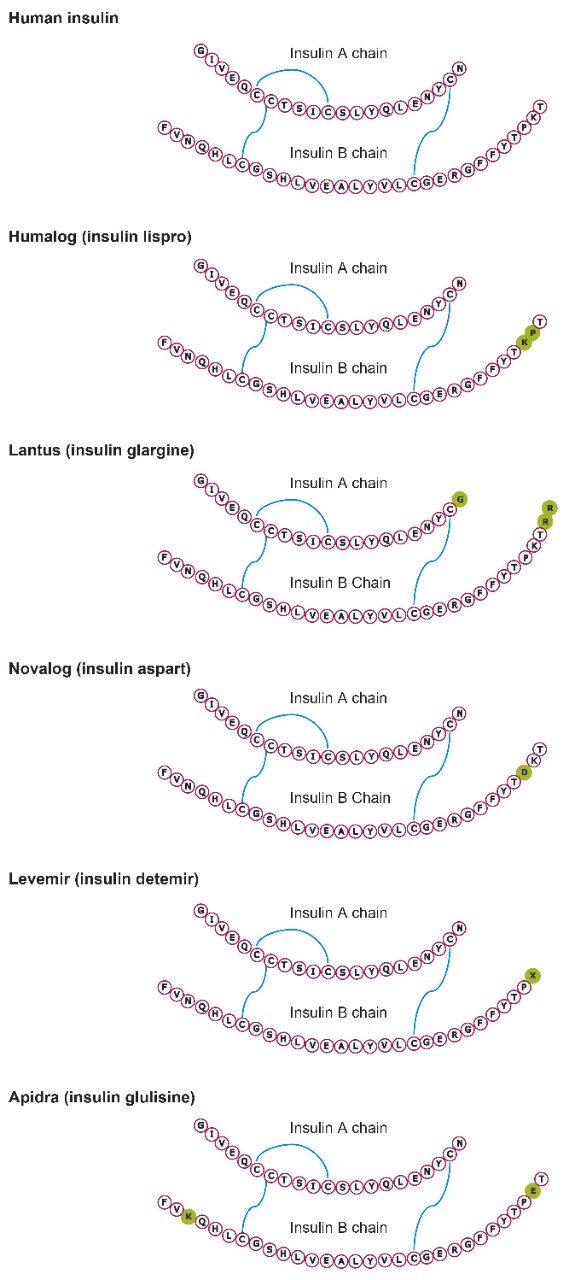
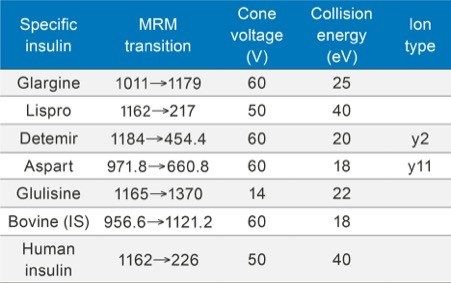
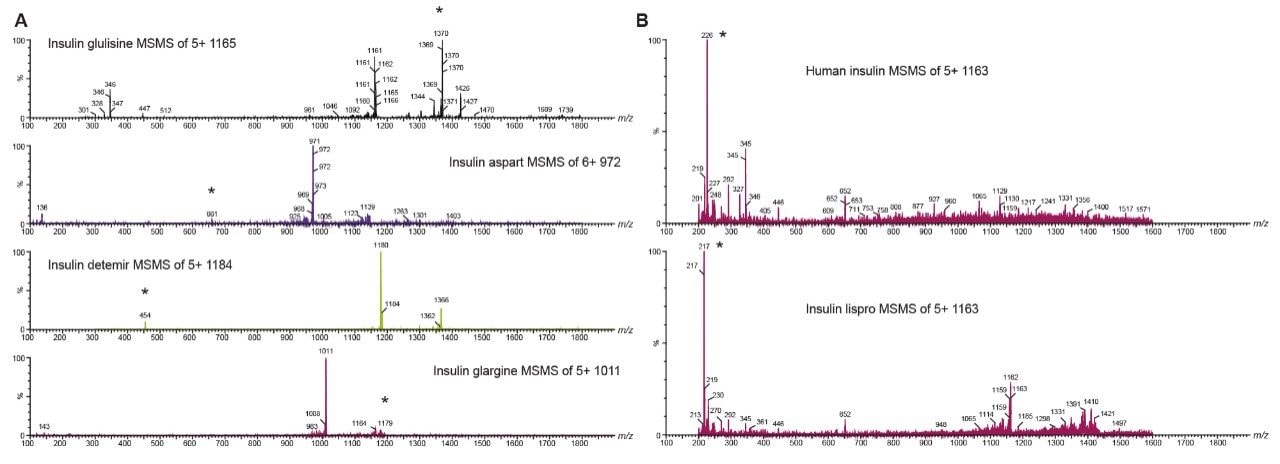
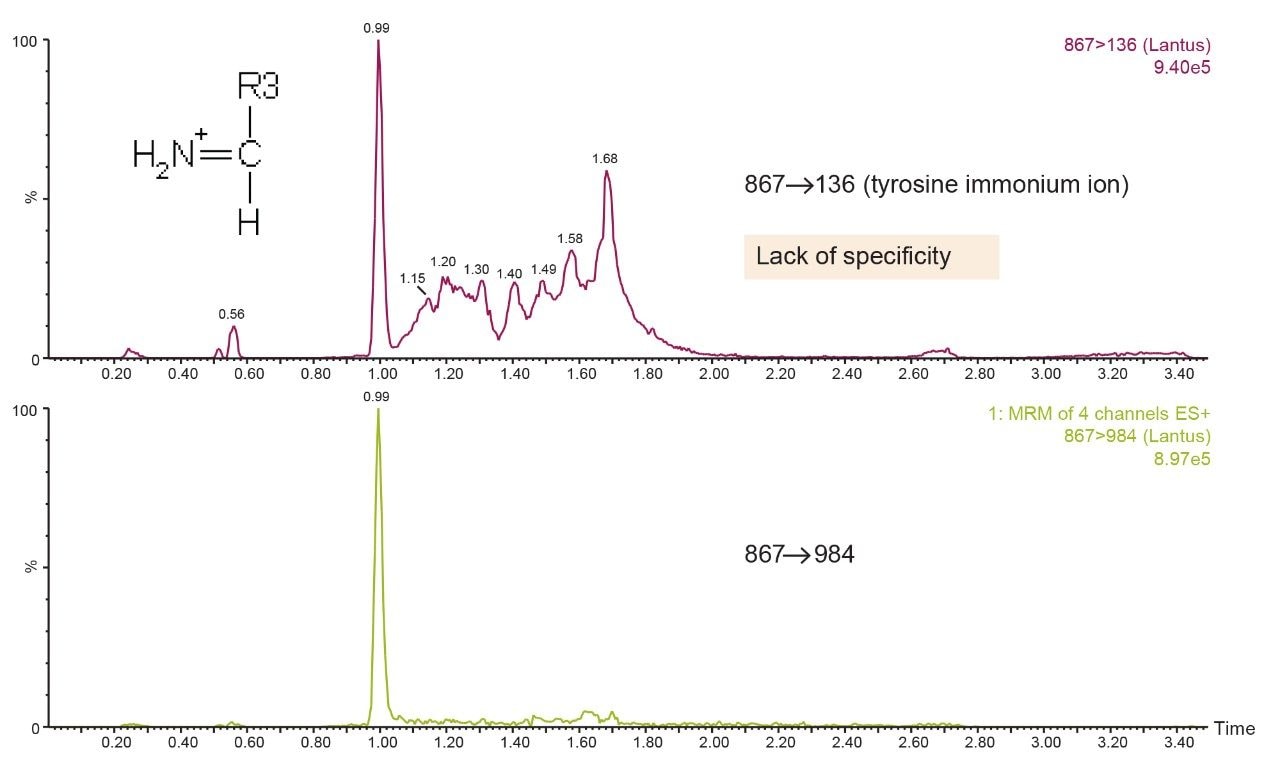
Insulin is perhaps one of the best known and earliest peptide therapeutics. Multiple long and fast-acting analogs have also been developed, and a patient may often be prescribed one of each for diabetes control. Quantification of biologics, such as insulins, has historically been performed using ligand binding assays (LBAs) such as ELISAS. LC-MS/MS, however, has certain advantages over LBAs, such as shorter development times, higher accuracy and precision, the ability to multiplex, no cross-reactivity, and the ability to readily distinguish between closely related insulins. Intact insulins are particularly difficult to analyze by LC-MS/MS, as MS sensitivity is low due to poor transfer into the gas phase, and poor fragmentation patterns exist due to the presence of multiple stabilizing disulfide bonds. In addition, insulin and its analogs suffer from non-specific binding and poor solubility, making LC and sample preparation method development difficult. A few LC-MS/MS methods do exist; however, most of those methods involve time-consuming and laborious immunoaffinity purification and/or nano-flow LC. Distinguishing between human insulin and insulin lispro (Humalog) is a very specific challenge for quantifying insulins, as they differ by a simple reversal in the position of two amino acids. Only a single low-molecular weight fragment differentiates the two, making selective sample preparation and chromatography critical.
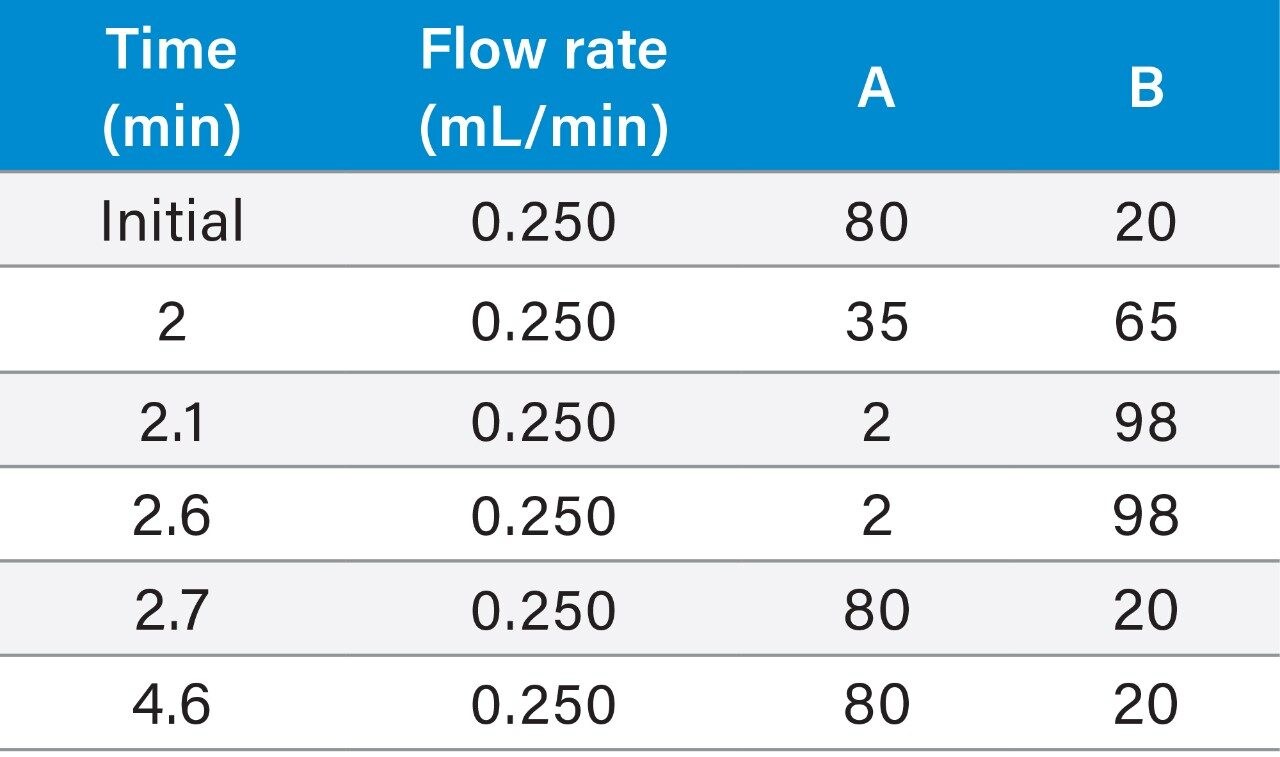
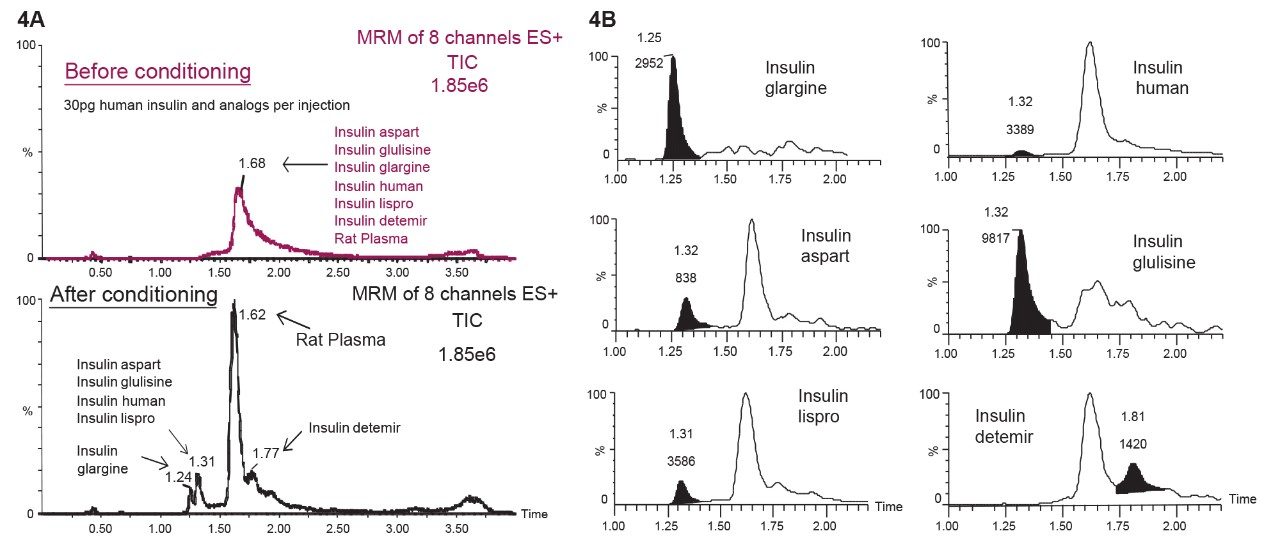
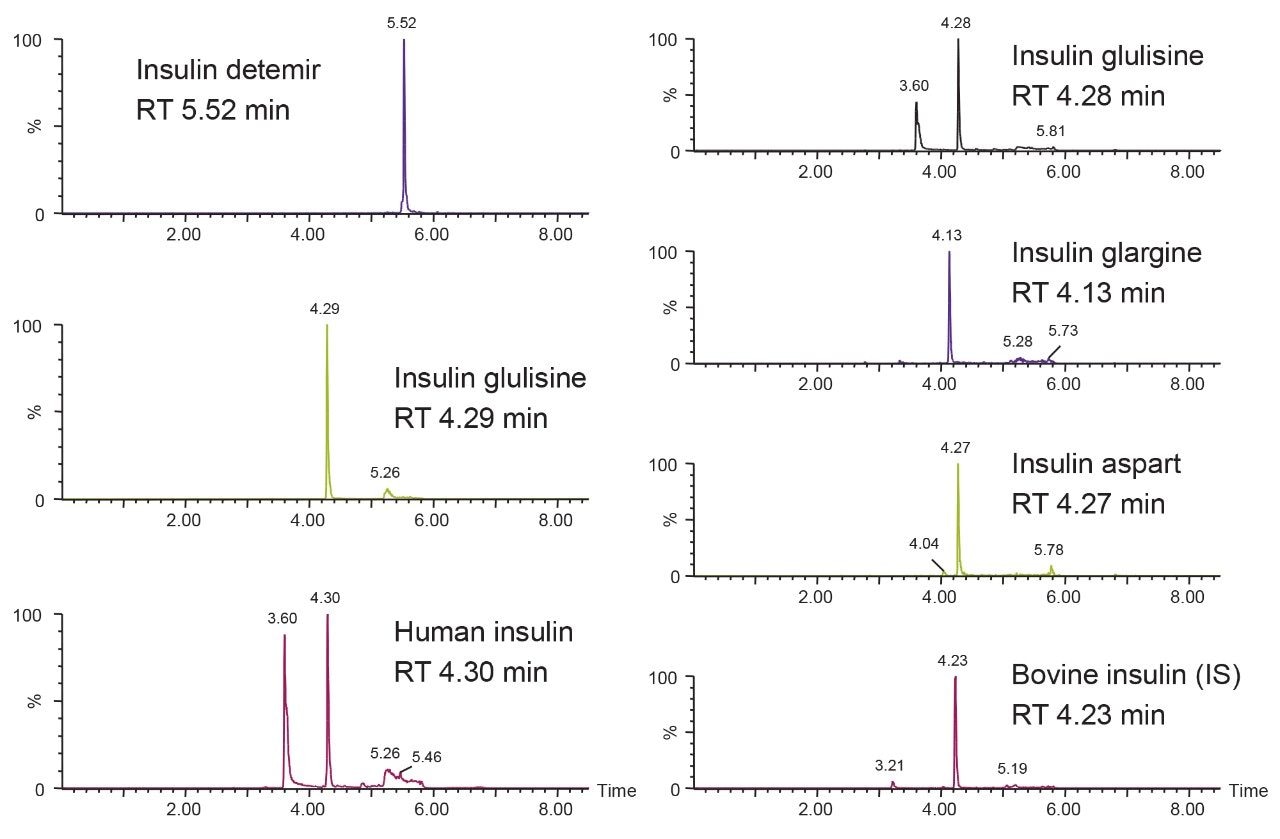
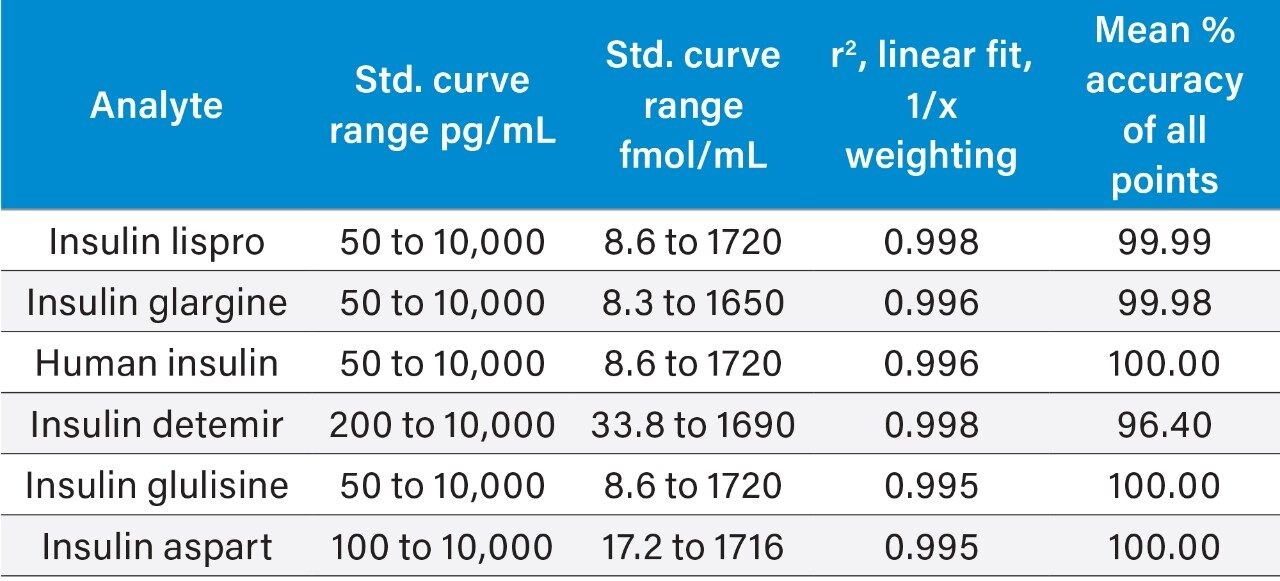
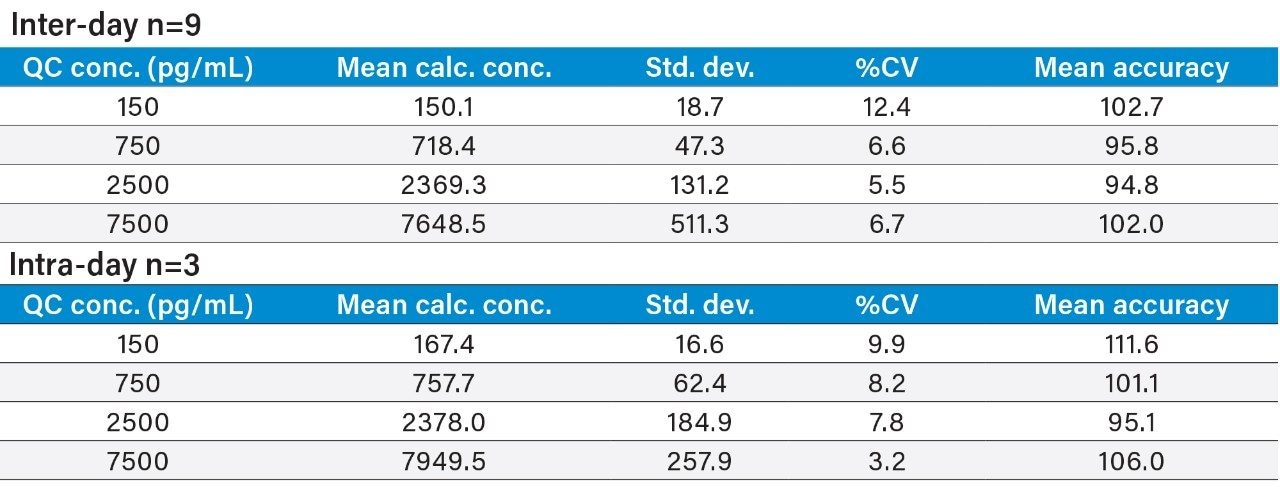
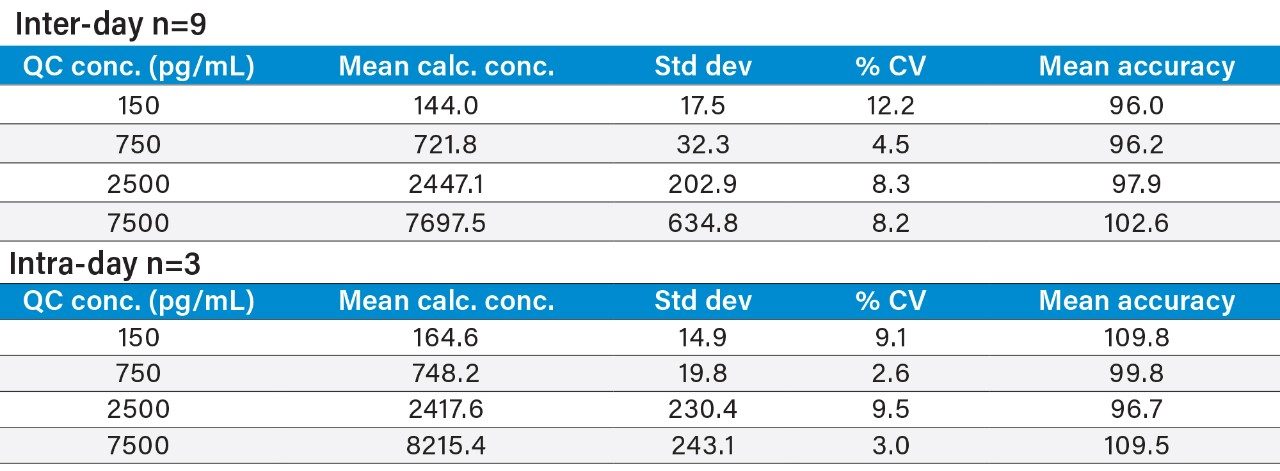
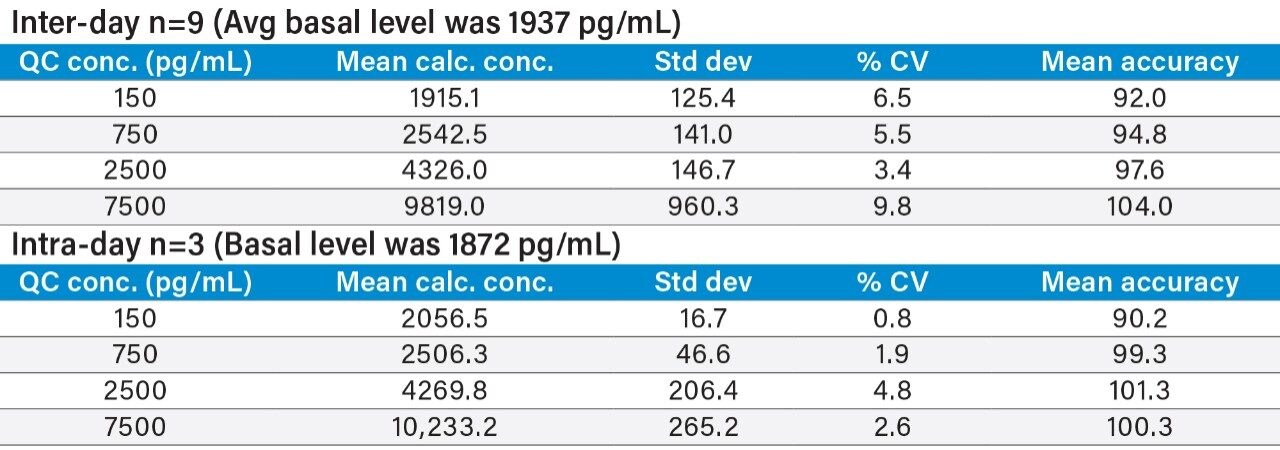
This study takes advantage of mixed-mode solid-phase extraction (SPE) and a high-efficiency, solid-core particle column that contains a low-level positive surface charge to eliminate interferences while facilitating high sensitivity quantification. Furthermore, selectivity studies show that the presence of high levels of human insulin, such as one might encounter in type II diabetic patients, does not interfere with quantification of lispro or any of the other analogs. This work provides a single, simple method for the simultaneous, direct quantification of intact human insulin and multiple insulin analogs in human plasma (Figure 1), achieving LODs of 50 to 200 pg/mL for each. Average accuracy for standard curve points was 99% to 100%. Average inter- and intra-day accuracies for QC samples were 98% and 94%, respectively. Average inter- and intra-day precisions for QC samples were 7.5% and 5.3%, respectively. Matrix factors for all analogs were calculated in six sources of human plasma and CVs of matrix factors were <15% in all cases, further supporting the selectivity of the method.