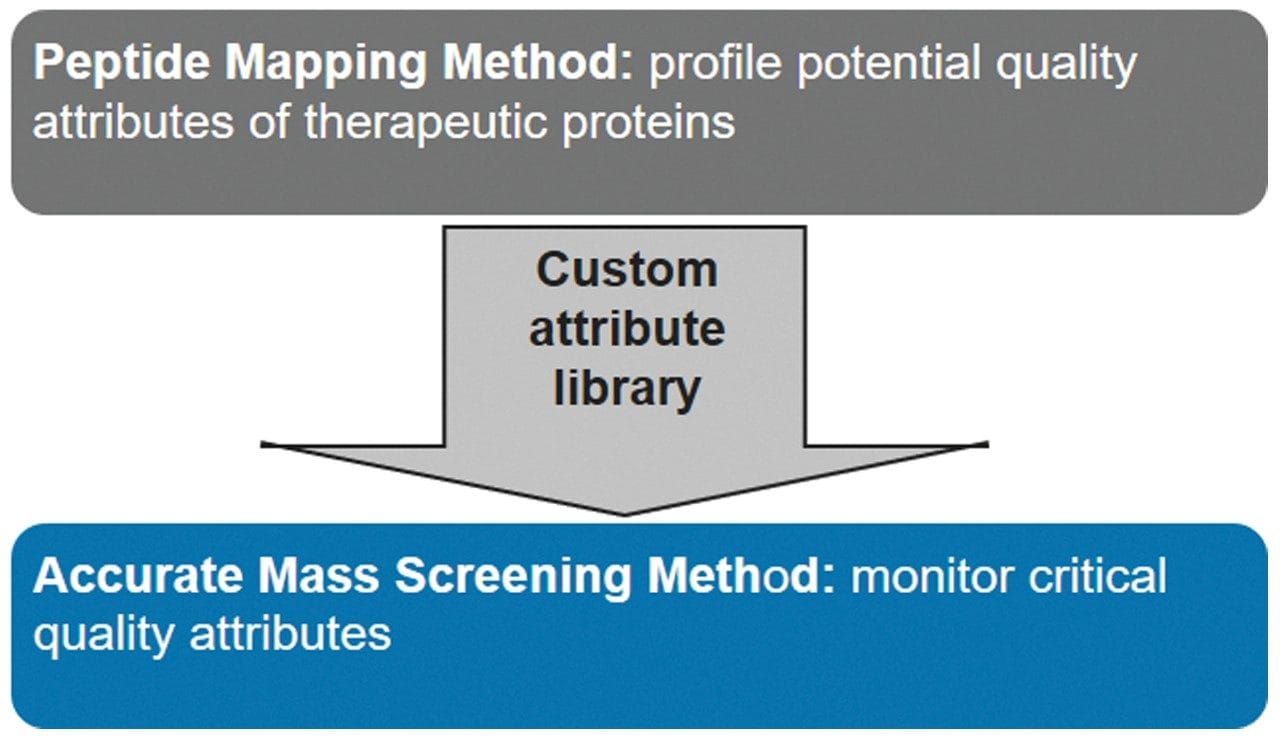
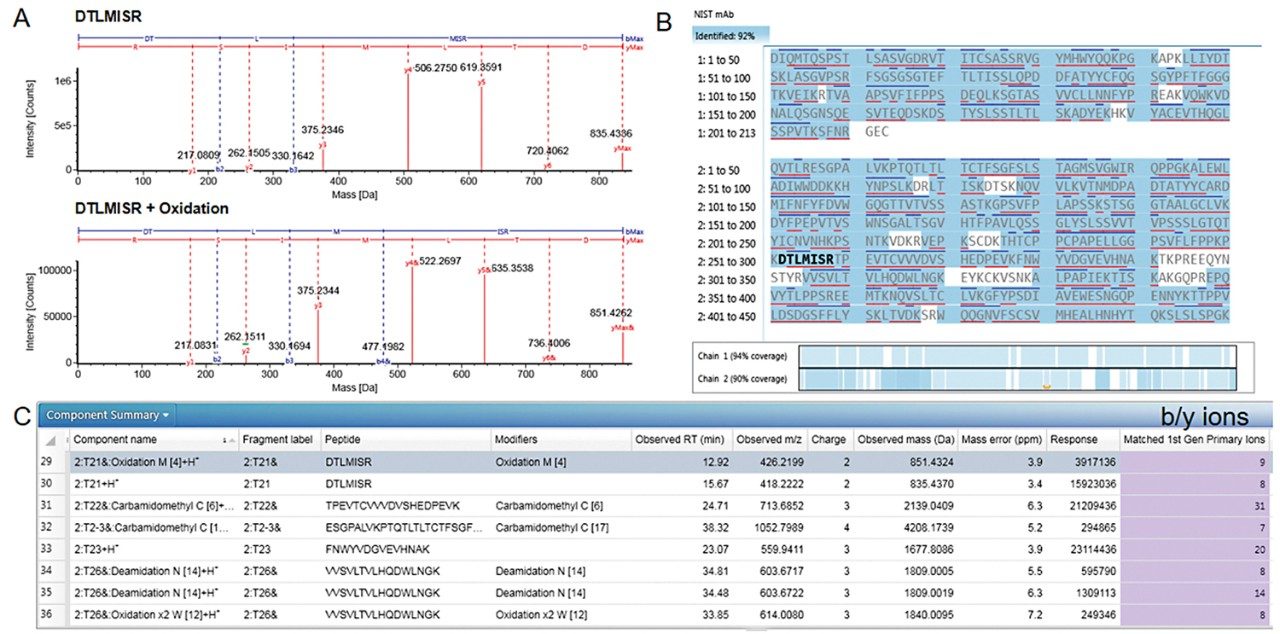
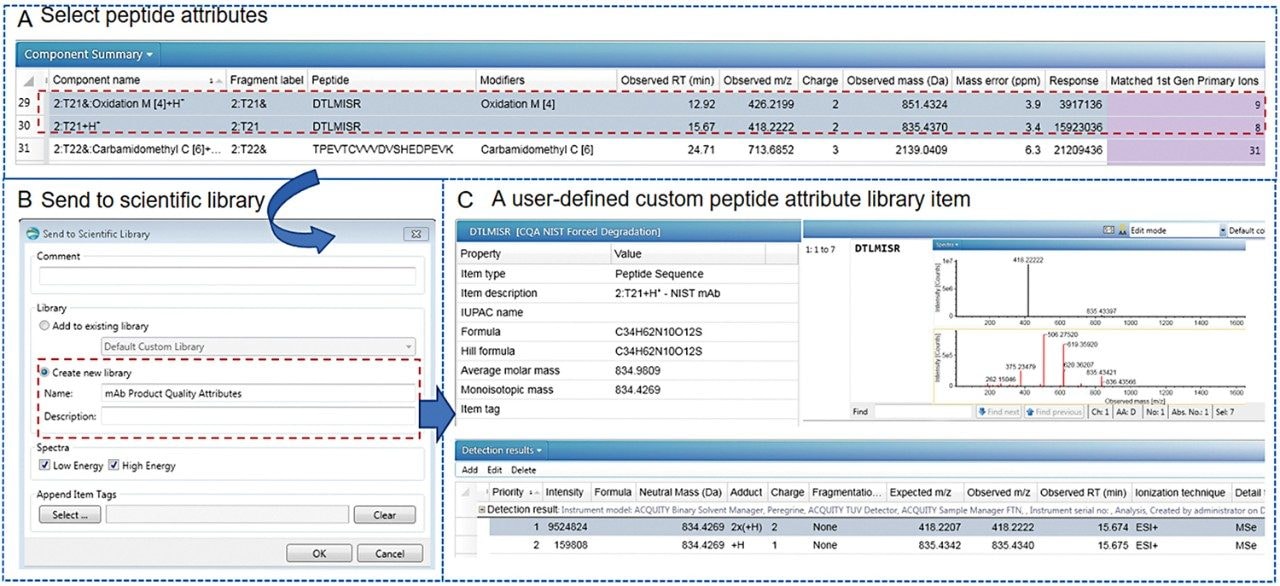
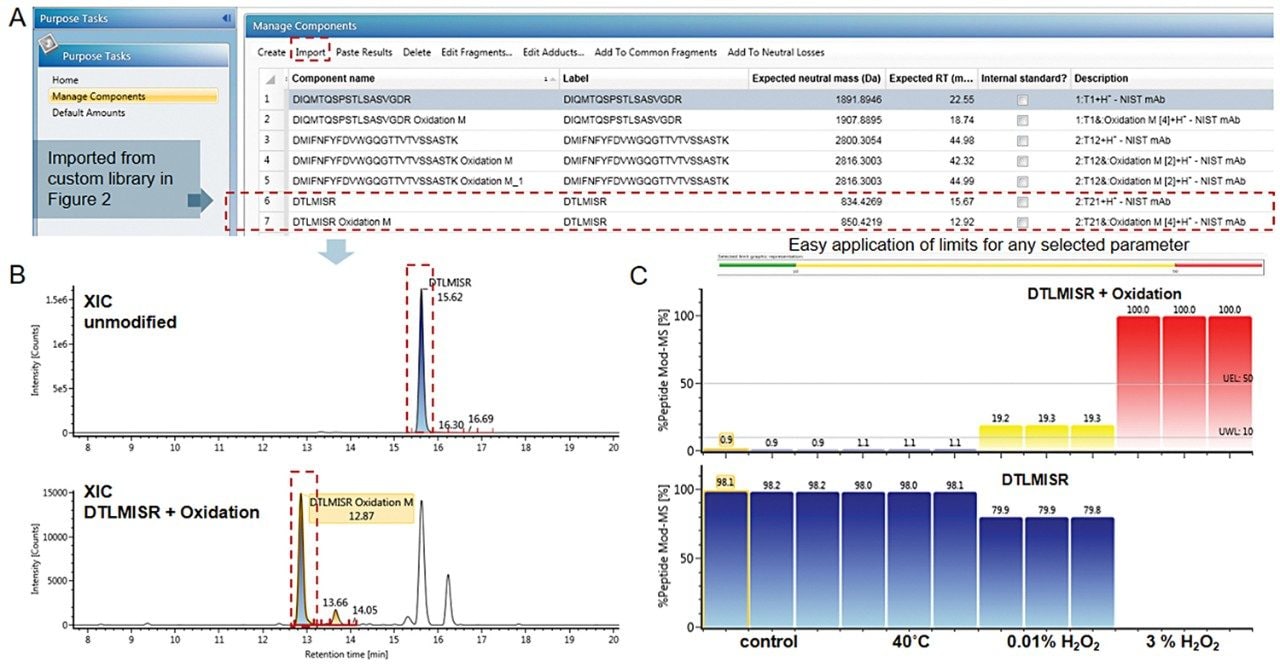
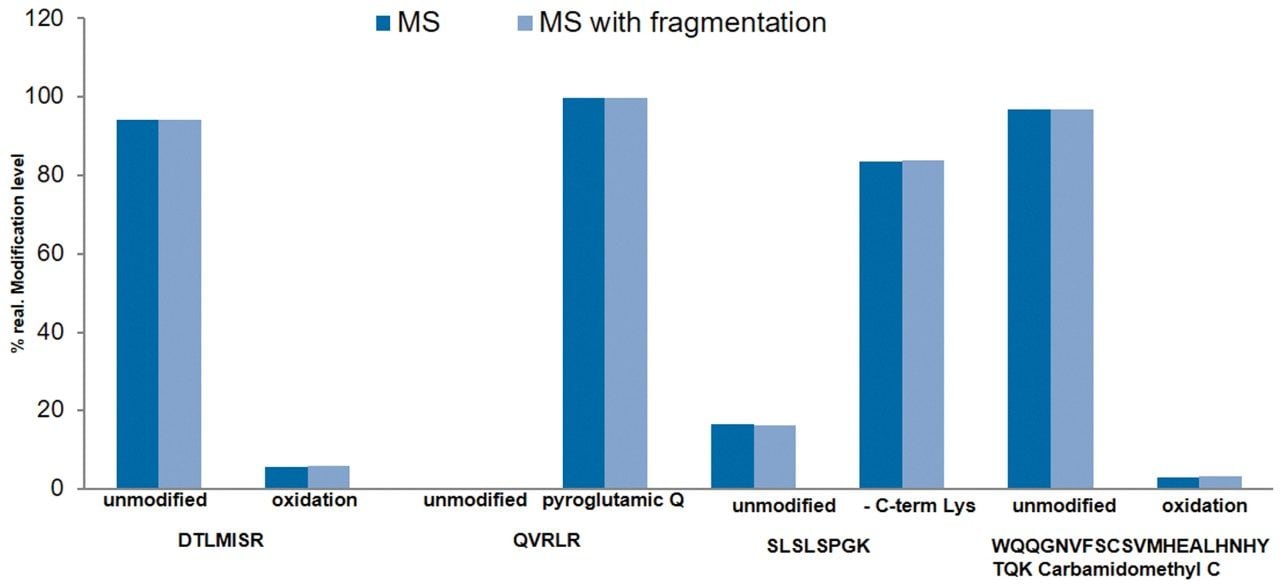
Biotherapeutics undergo rigorous characterization and monitoring during their development and manufacturing to establish product quality and safety for regulatory compliance.1,2 High-performance LC-MS technology, often utilized for the characterization of a protein’s chemical and post-translational modifications at the peptide level, has now been expanded beyond the conventional realm to attribute monitoring across different stages of the product life cycle. Routine monitoring of these quality attributes requires fit-for-purpose analytical platforms that are robust, easy to operate, and readily deployable in laboratories across an organization. The BioAccord System is a new, compact, high-performance, LC-MS system purposefully designed to meet such requirements. It consists of an ACQUITY UPLC I-Class PLUS, ACQUITY RDa Detector featuring SmartMS Technology, compliance-ready UNIFI Software, and consumables all tailored to assist biopharmaceutical applications. The UNIFI Scientific Information System provides integrated, analytical workflow methods with automated data acquisition, processing and reporting capabilities allowing peptide monitoring assays to be readily performed and documented for a single or large batch of samples. In this application note, we demonstrate peptide mapping and monitoring capabilities of the BioAccord System using forced degradation3,4 study of a monoclonal antibody (mAb) as a case study.