Nitrofurans (NFs) are a group of broad spectrum antibiotics. Due to health concerns, nitrofurans are now prohibited for use in food-producing animals in most jurisdictions. They are still authorized for human medicine and for the treatment of non-food animals. Nitrofurans are widely manufactured, sold, and hence available for misuse.1
There have been frequent findings of nitrofuran residues in honey, poultry, and aquaculture products imported to EU countries, which has led to product recalls, border rejections, and de-listed suppliers. Violations have resulted in implementation of emergency measures that have necessitated mandatory pre-export testing (PET), widespread voluntary pre-harvest tests (PHT), and an increase both in the analysis of imports at border control within the EU and in the frequency of European Commission Food and Veterinary Office (FVO) visits.2
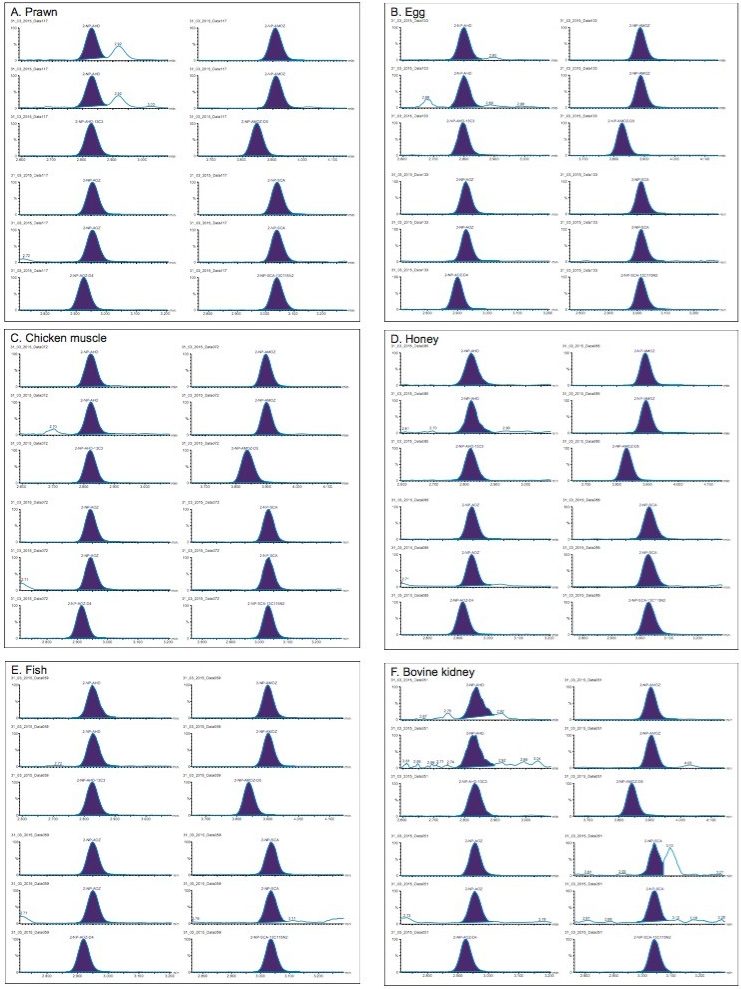
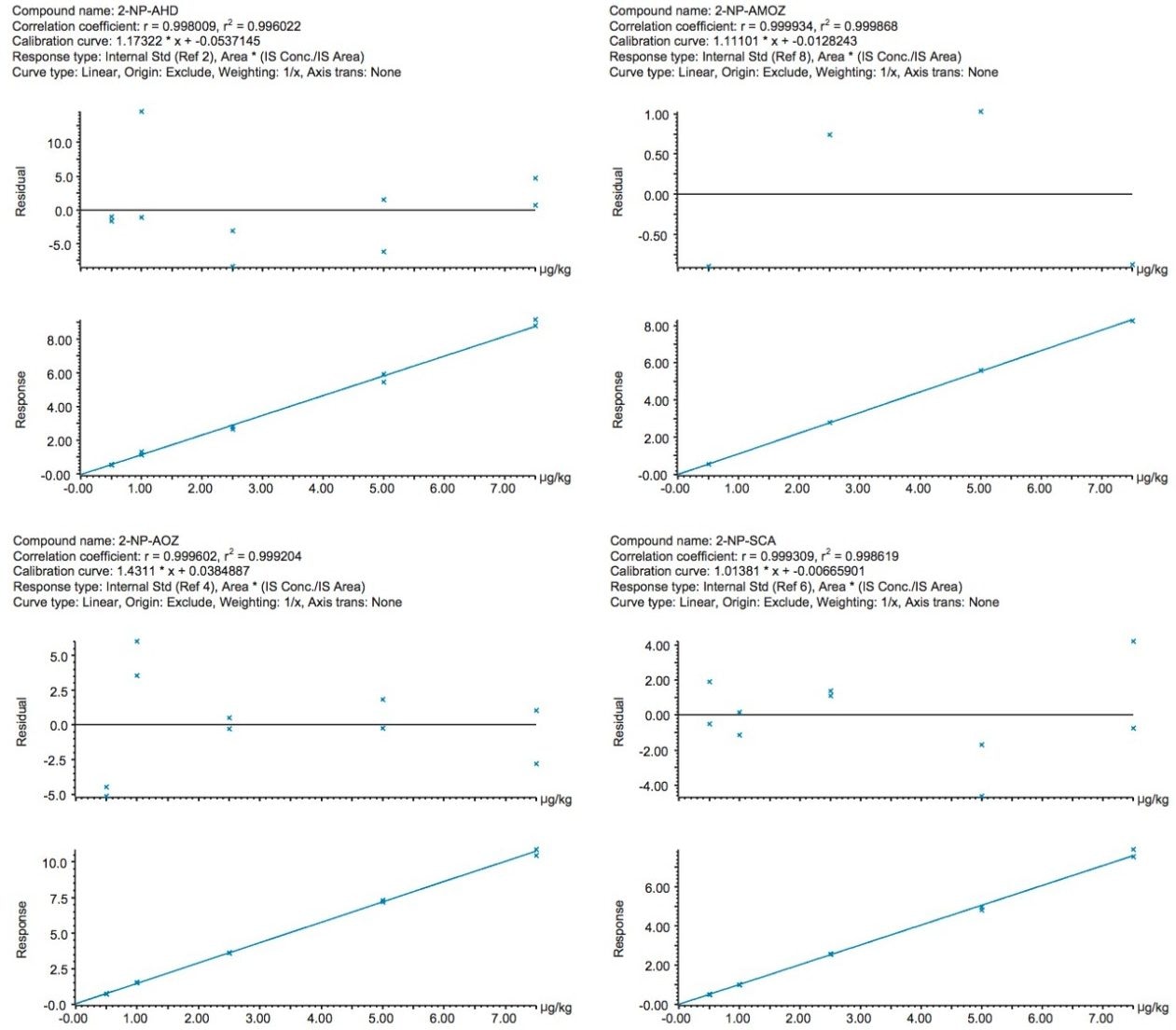
The EU Minimum Required Performance Limit (MRPL) in poultry meat and aquaculture is 1 µg/kg for each of the four nitrofurans, measured as their respective tissue-bound metabolites (EU Commission Decision 2003).3 MRPLs are ‘the minimum content of an analyte in a sample, which at least has to be detected and confirmed’. They are also the reference point for action (Action Levels) when evaluating food consignments. Laboratories must demonstrate that their calculated Detection Capability (CCβ) and Decision Limit (CCα) values are at or below the MRPL.4 Although enforcement action is only taken when a residue exceeds the MRPL, non-compliant samples below the MRPL must still be monitored. Suppliers and importers can set even lower limits for PET based upon trading decisions to provide better warranties to their customers and to gain commercial advantage. Meeting these requirements requires the continued development of highly sensitive and specific analytical methodology based upon UPLC-MS/MS.
Previous studies have demonstrated that parent nitrofurans deplete rapidly in animals and that they are extensively metabolized to tissue-bound metabolites.5 Methods have been described for various animal tissues such as kidney for official control, muscle, honey, shrimp, eggs, and milk for assessing risk to the consumer. Parent nitrofurans are only sought in medicated feeds used for animal production and aquaculture.
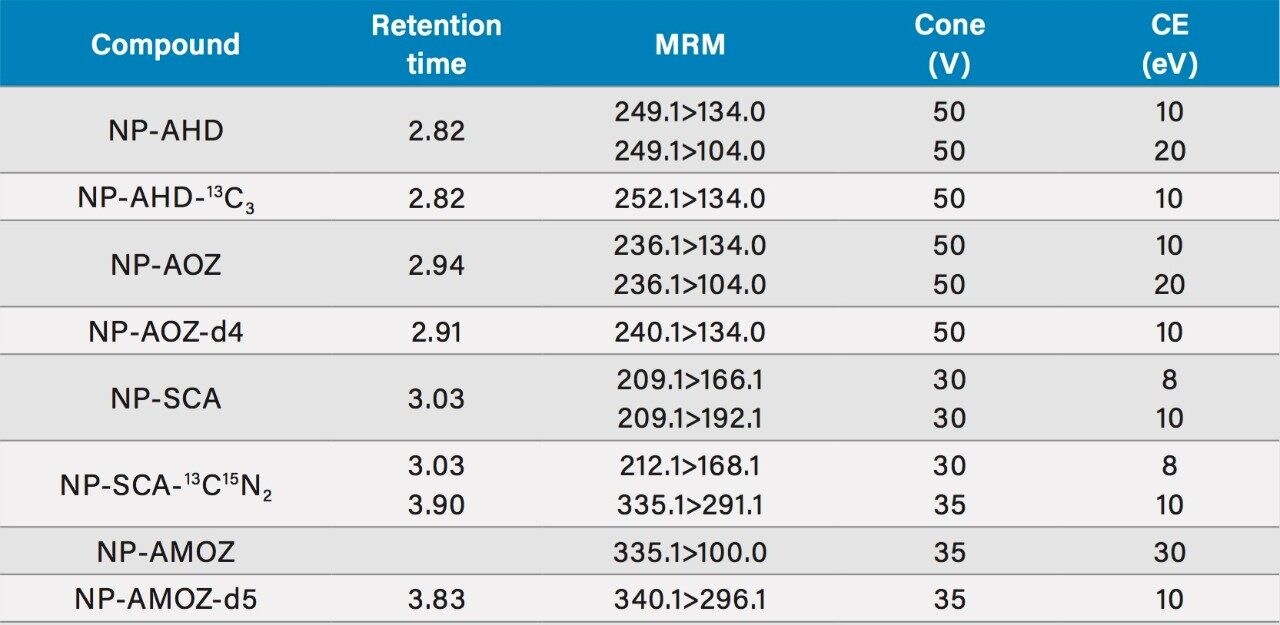
Commonly sought parent nitrofurans and associated metabolites include: furazolidone as 3-amino-2-oxazolidinone (AOZ), nitrofurazone as semicarbazide (SCA), furaltadone as 3-amino-5-morpholinomethyl-2-oxazolidinone (AMOZ) and nitrofurantoin as 1-aminohydantoin (AHD).
Derivatization improves extraction efficiency from the acidic aqueous phase into the organic solvent, imparts functionality amenable to reversed-phase chromatography for both SPE and UPLC, and improves electrospray ionization efficiency.