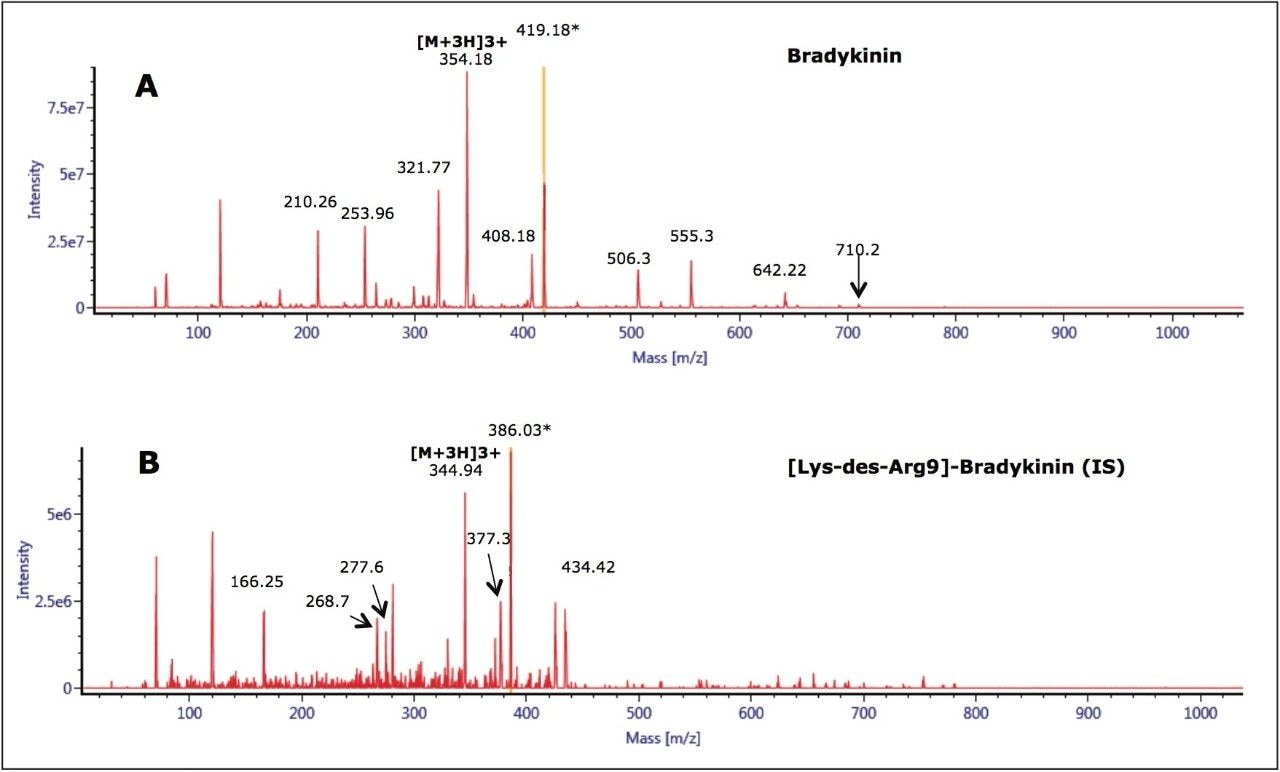
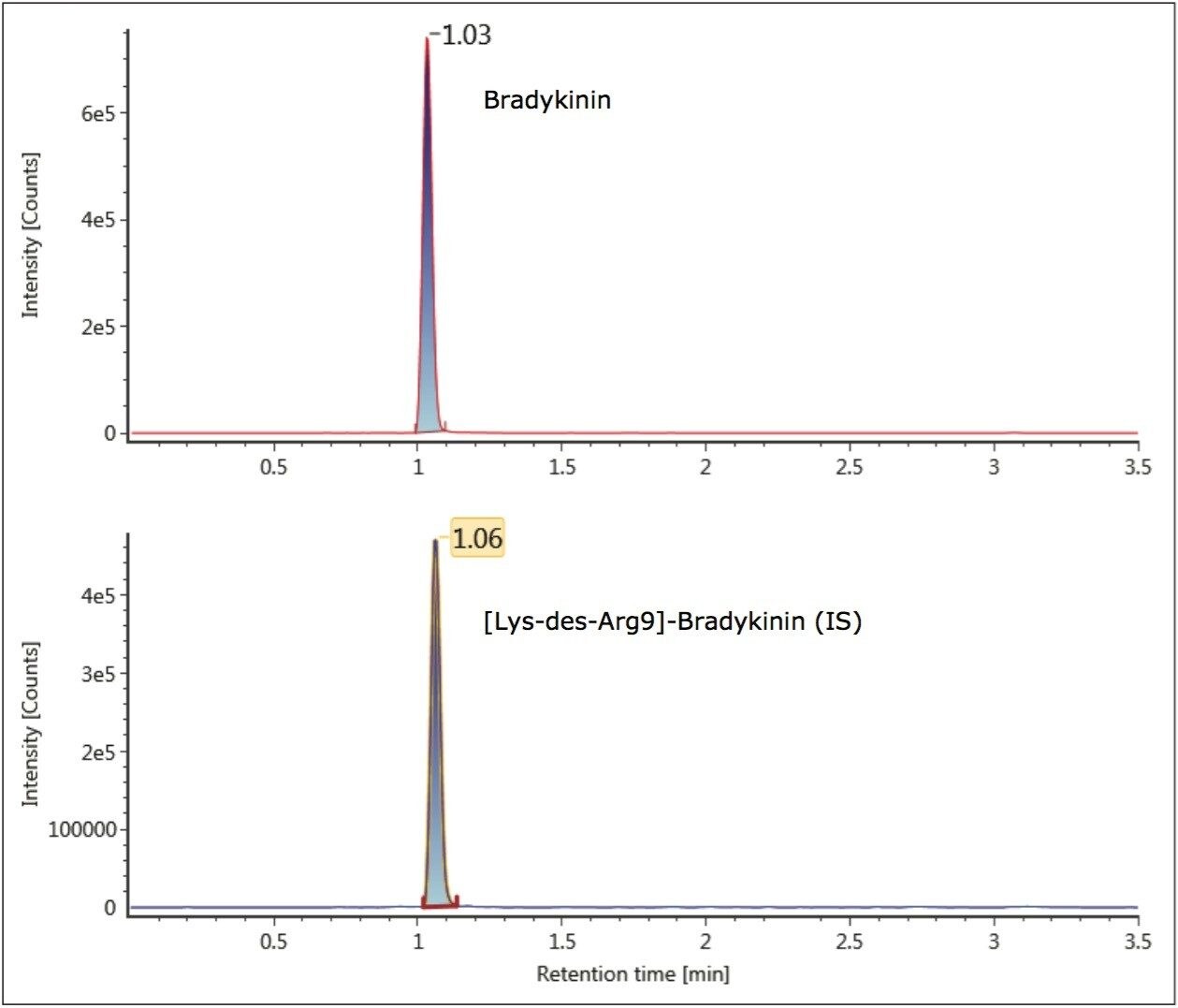
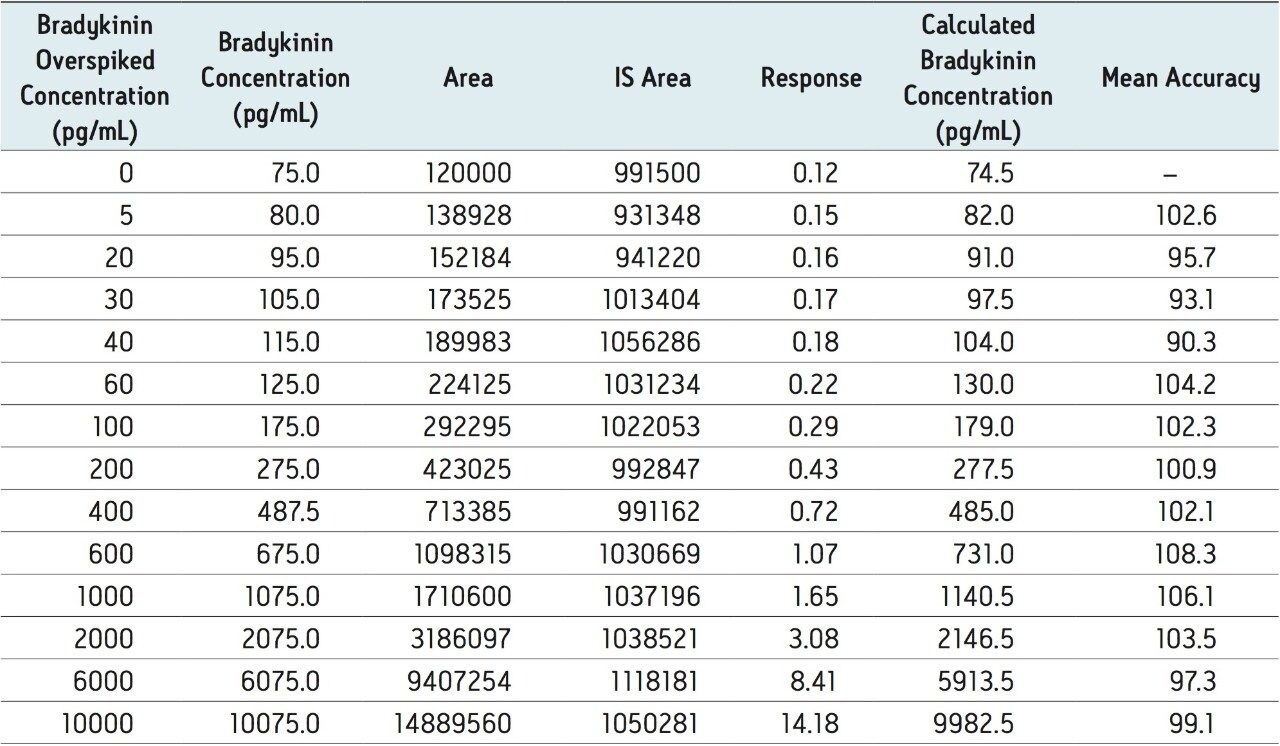
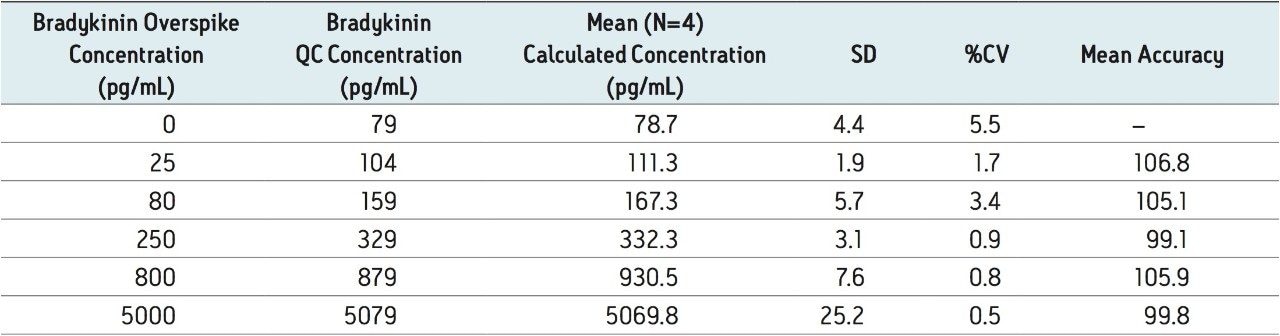
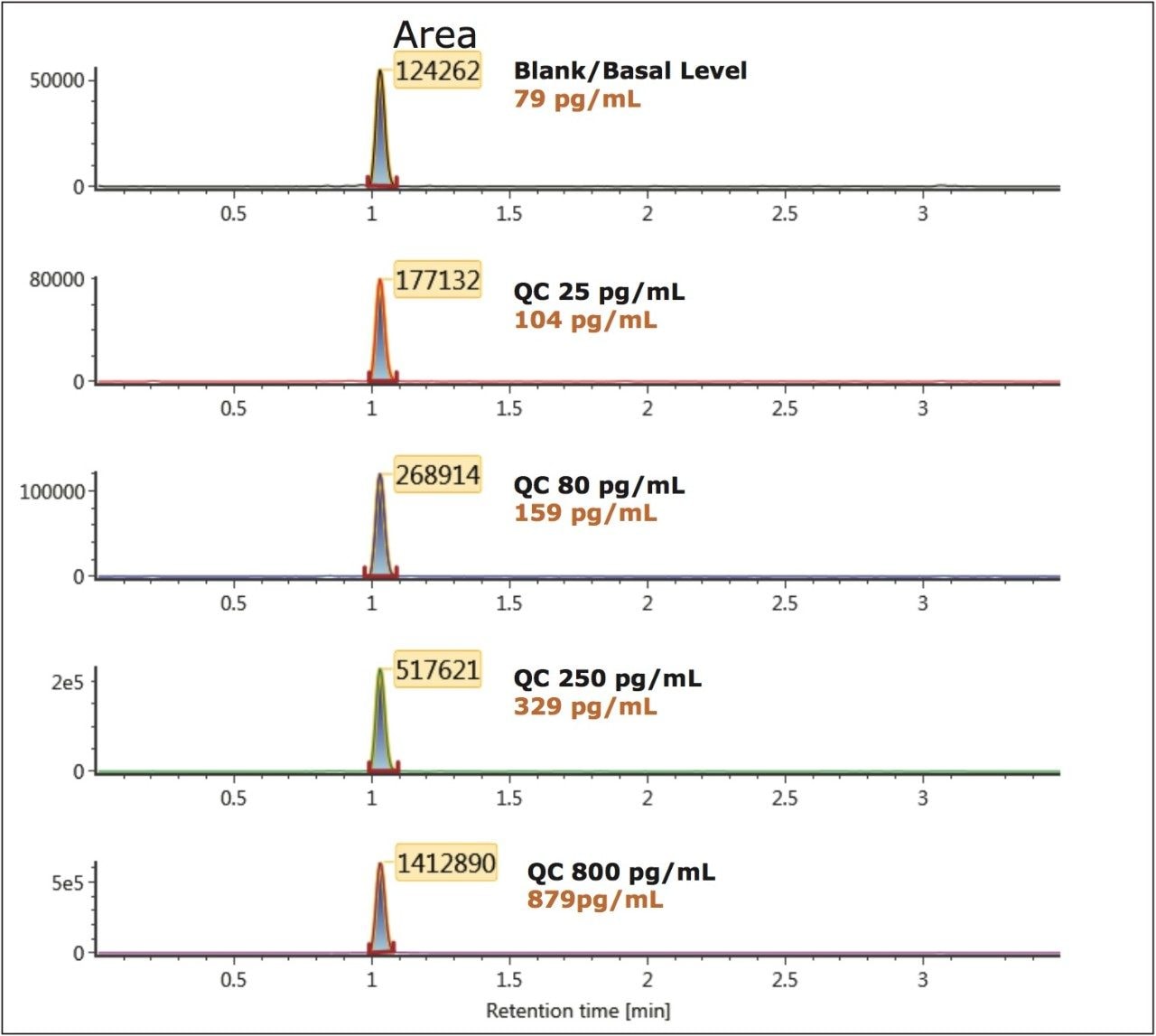
Linearity, accuracy, and precision
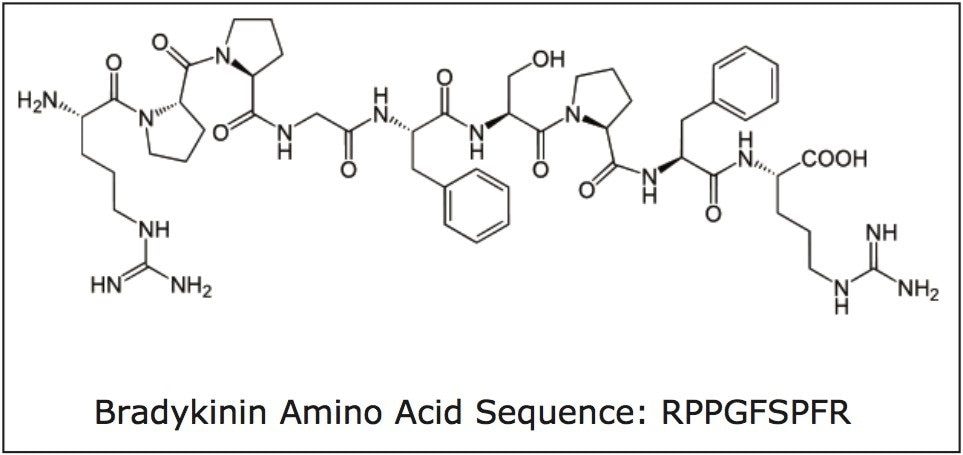
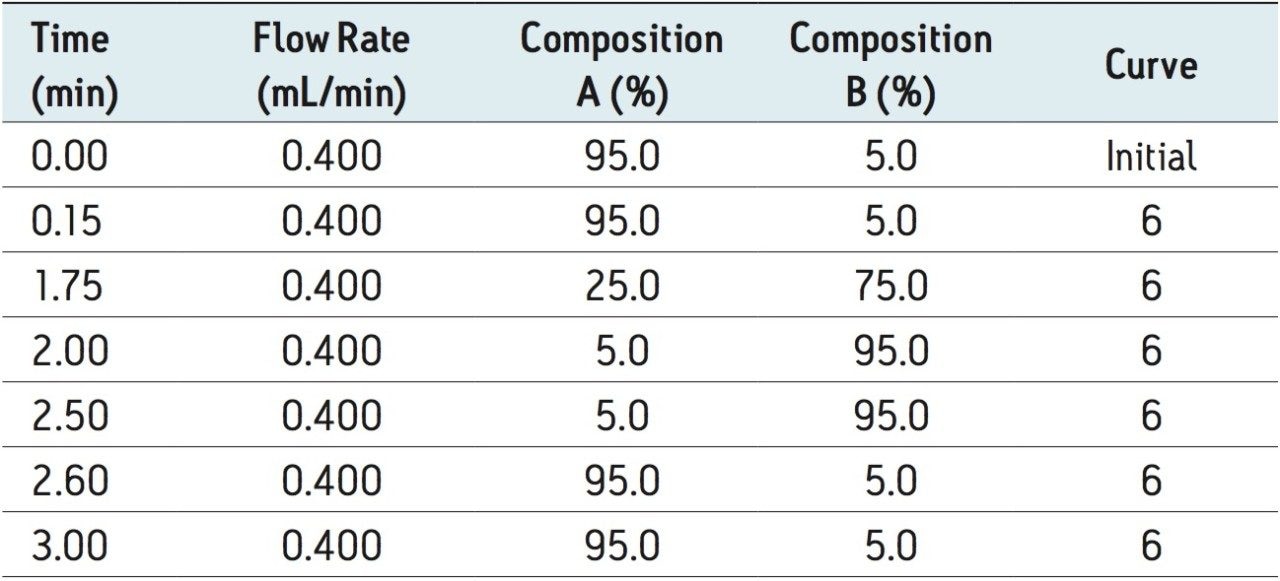
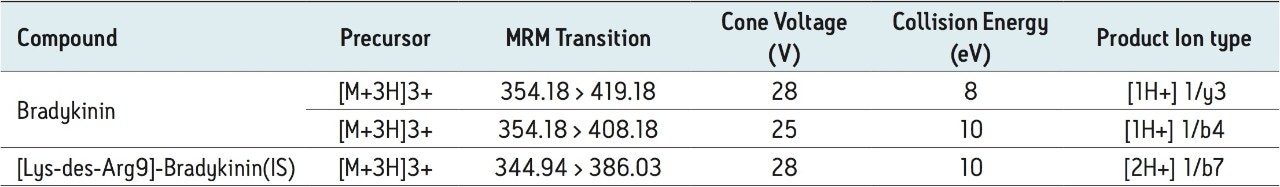
To generate standard curves, human plasma was fortified with bradykinin at the following final concentrations: 5, 20, 30, 60, 100, 200, 400, 600, 1000, 2000, 6000, and 10000 pg/mL. Quality control (QC) samples were prepared in human plasma at the following concentrations: 25, 80, 250, 800, and 5000 pg/mL. [Lys-des-Arg9] Bradykinin (final concentration of 0.5 ng/mL) was used as the internal standard (IS) for bradykinin. Peak area ratios (PARs) of the analyte peak area to the IS peak were calculated. The calibration curve, prepared in human plasma, was constructed using PARs of the calibration samples by applying a one/ concentration (1/x) weighted linear regression model. All QC sample concentrations were then calculated from their PARS against the calibration curve. Due to the presence of endogenous bradykinin, standard addition was used. The mean basal level of bradykinin in control plasma samples was determined by calculating the x-intercept. The calculated basal level of bradykinin was then added to the spiked concentration for all standard curve and QC samples to enable accurate quantification. Using 1/x regression, bradykinin was linear with an R2 value of >0.99. A summary of standard curve performance is shown in is shown in Table 3. Results from QC analysis are shown in Table 4, and representative chromatograms of endogenous bradykinin and QC’s are shown in Figure 5. QC samples, at all levels demonstrated very good accuracy and precision, easily meeting recommended FDA acceptance criteria outlined in the white papers describing best practices in bioanalytical method validation for LC-MS/MS assays.4,5 Mean endogenous basal levels of bradykinin were determined to be 79 pg/mL in human plasma with a SD and %CV of 4.4 and 5.5, respectively. This indicates a robust and reproducible method.