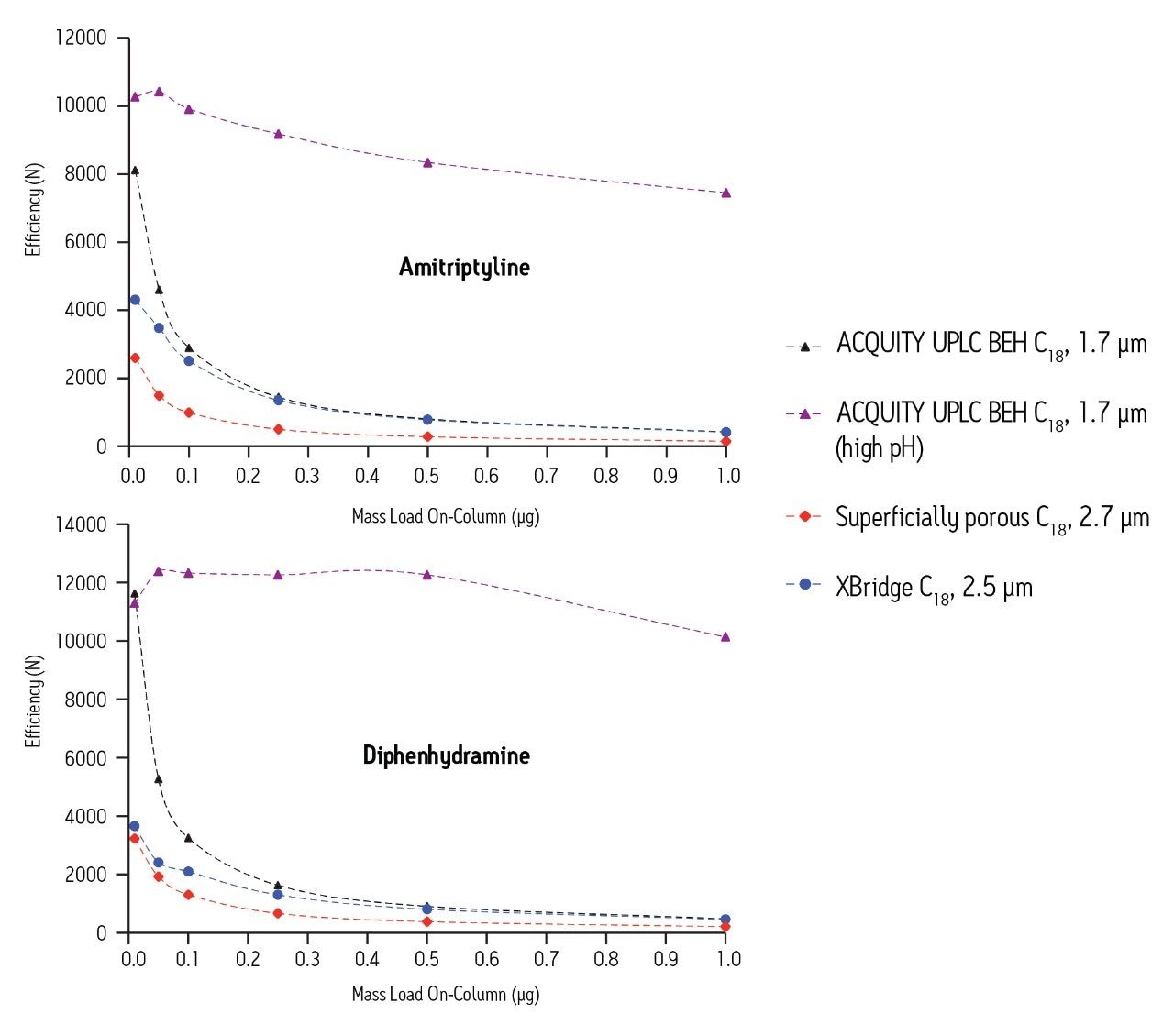
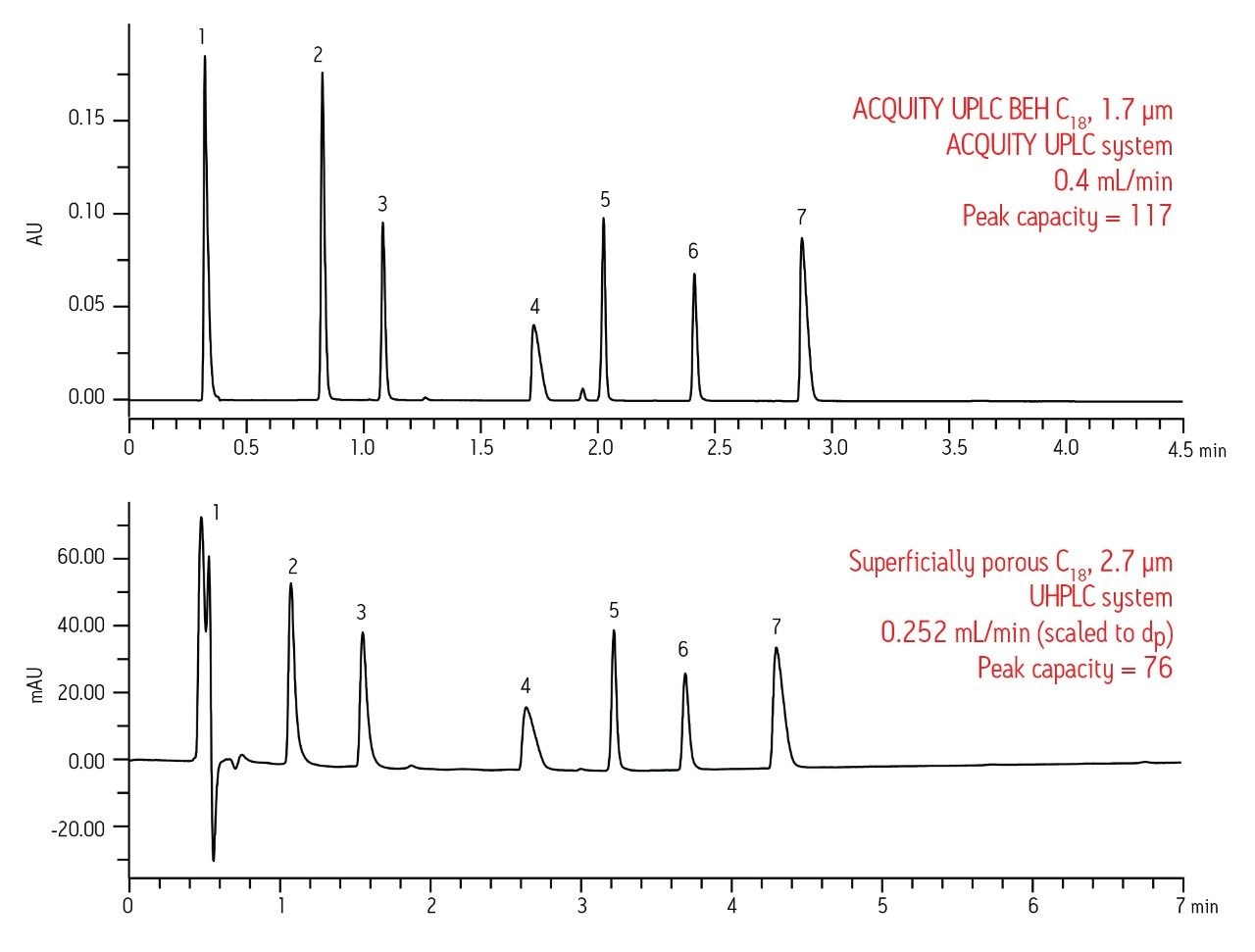
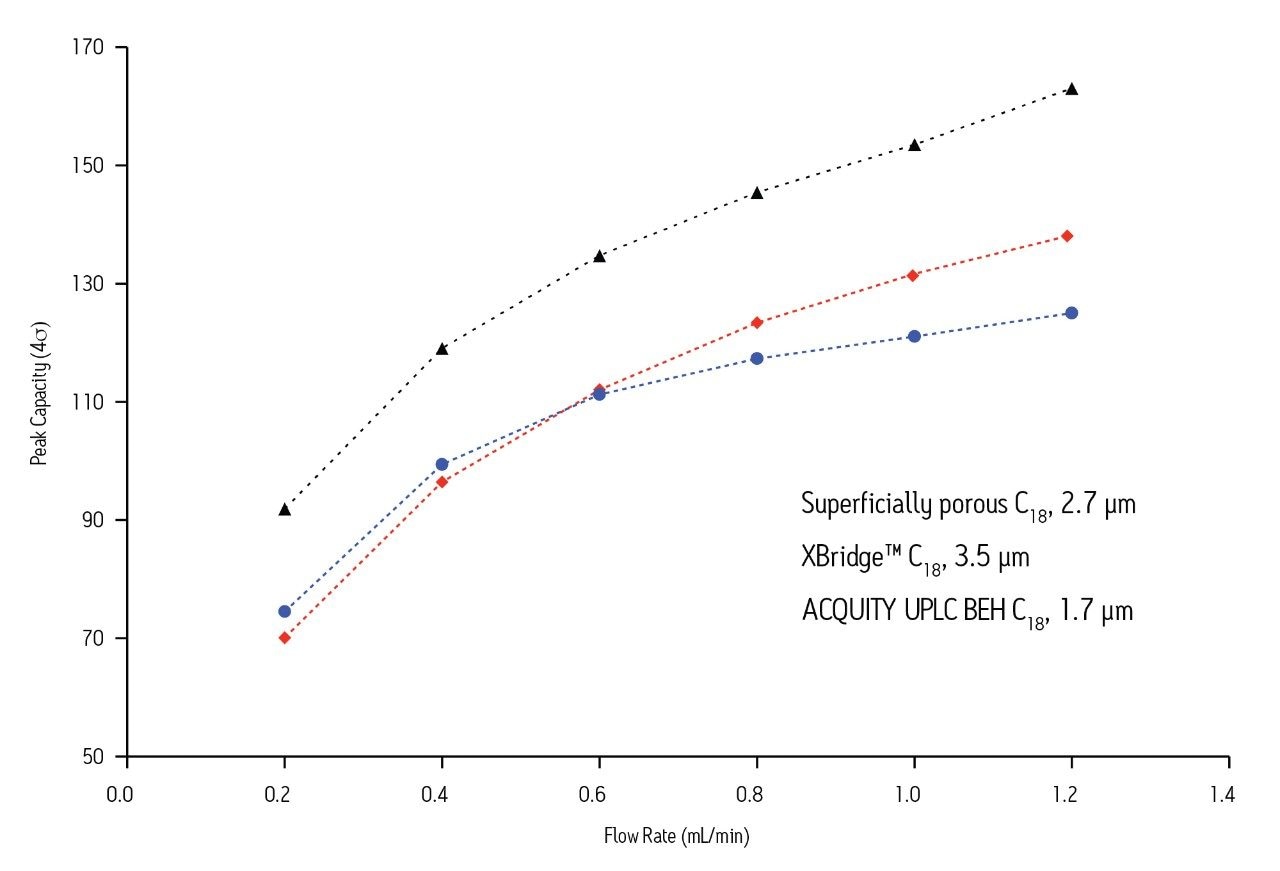
The reasons for these differences in performance are numerous. First, as shown in Figure 1, superficially-porous particle columns become overloaded, even at very low mass loads for basic compounds, whereas fully-porous particles do not overload as quickly. Second, in order to achieve the low gradient delay volume on the UHPLC system, certain components of the system must be by-passed, including the pump pulse dampener, injection loop, etc. This is most likely the reason for the disturbances in the baseline seen early in the UHPLC chromatogram. In order to achieve the low system delay volume on the UHPLC system, the sample loop is taken off-line after the injection. It is likely that the baseline disturbances seen in the UHPLC separation are a result of the injector valve cycling between the load and inject positions during this process. Finally, there are differences between the detector cells and settings of the two systems.
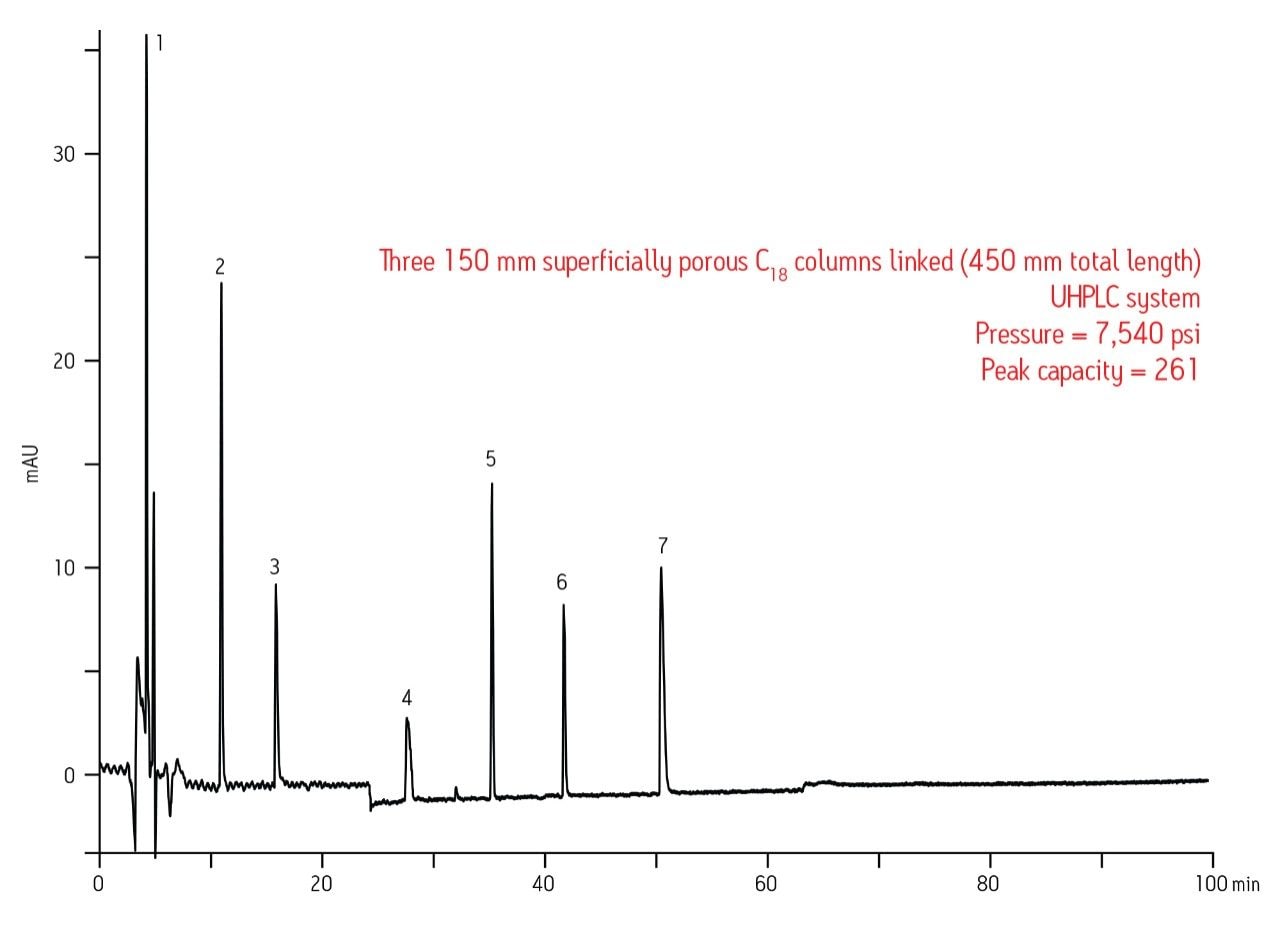
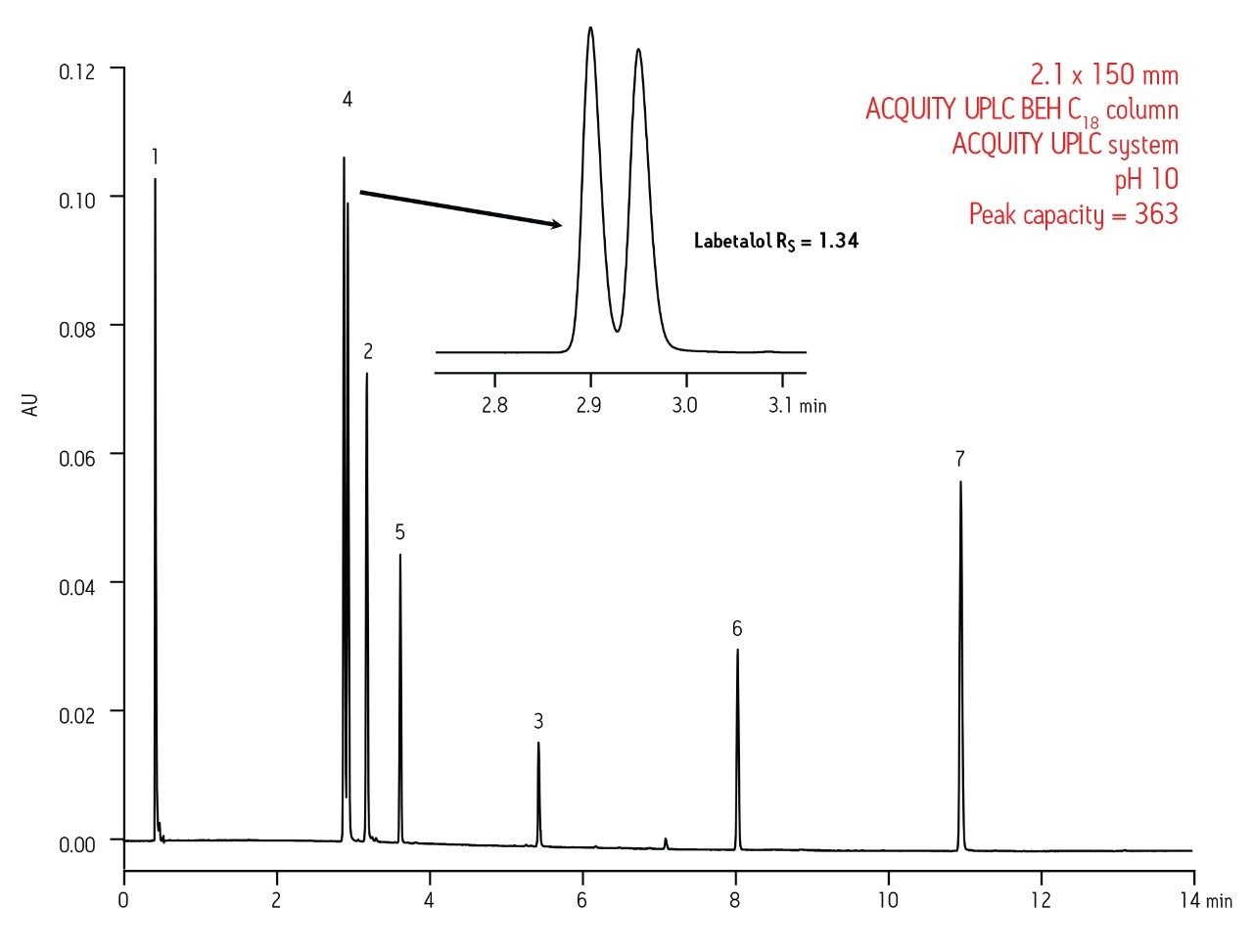
Since the instrument and column have to work together to produce high quality data, sacrificing the performance of one will ultimately lead to sub-optimal performance of the entire system. For the data shown in Figure 2, it is clear that better separation performance can be achieved with UPLC when compared to UHPLC, especially for basic drugs. This was confirmed by running the 1.7 μm fully-porous particle column on the UHPLC system under the same conditions and observing a 38% loss in peak capacity, which is simply due to the contributions of the chromatographic system (data not shown).