This new FastDDA algorithm includes the following features:
- Acquisition rates up to 30 MS/MS spectra per second
- Precursor selection (and exclusion) by charge state
- Rapid decision making, made during inter-scan delay
- New iTRAQ MS/MS function
- Inclusion list trigger based upon accurate mass of precursor ions
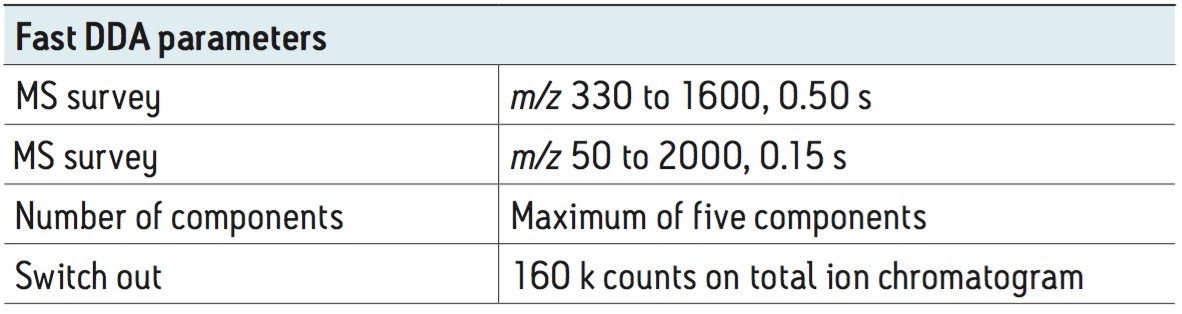
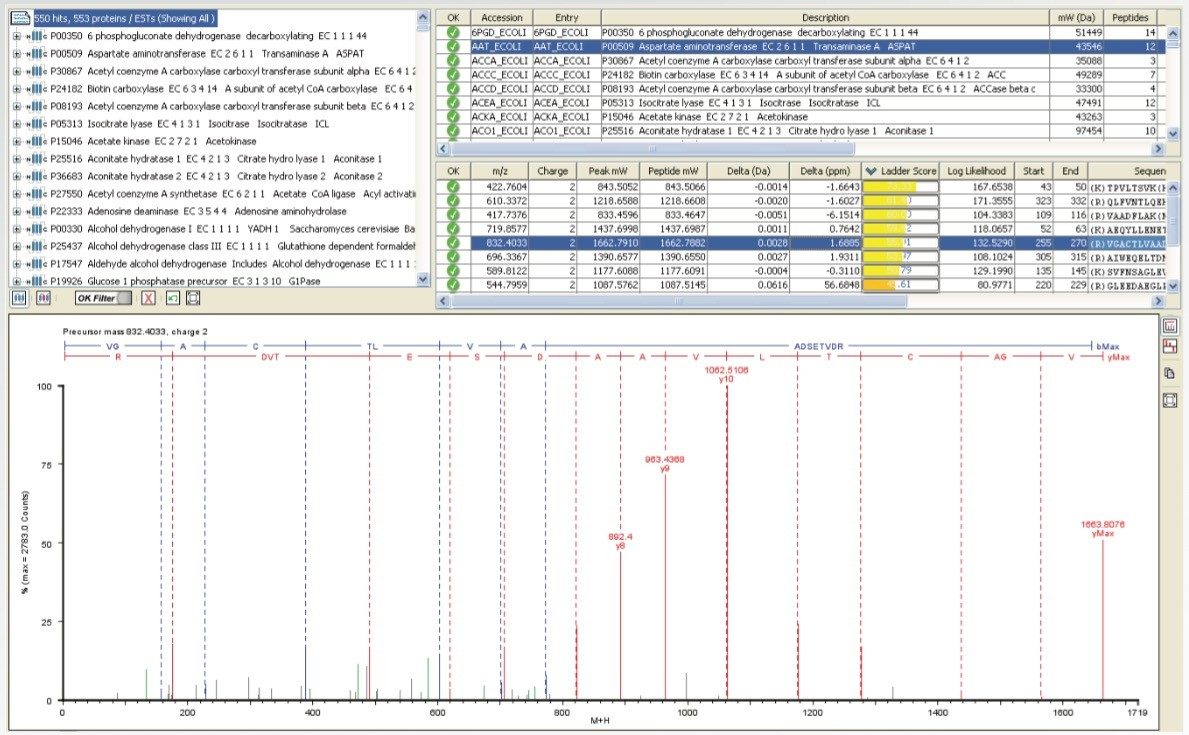
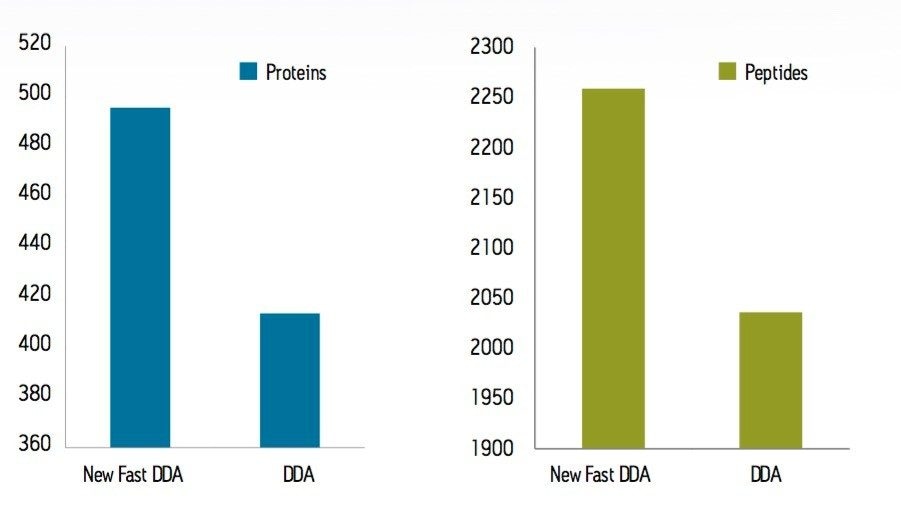
To test the performance of the new algorithm, a complex tryptic digest from the cytosolic fraction of E.coli was separated on a nanoACQUITY UPLC System with an ACQUITY UPLC BEH 1.7 µM, 75 µM x 100 mm Column. A gradient from 1% to 40% acetonitrile + 0.1% formic acid over 90 minutes was used at a flow rate of 300 nL/min. The UPLC eluent was passed directly into the NanoFlow ion source of a Xevo G2 QTof Mass Spectrometer. For all experiments, 400 ng of total protein digest were injected on column, and the FastDDA parameters that were used in these experiments are shown in Table 1. The results in terms of peptides and proteins identified for the FastDDA algorithm are shown in Figure 1. The results obtained from the FastDDA code were then directly compared to the previous MassLynx Software DDA acquisition code, as shown in Figure 2.
It can clearly be seen that both the number of peptides and proteins identified by the new FastDDA algorithm is superior.