The following sections will detail the practical utility of these new workflows for biotherapeutic peptide mapping analysis.
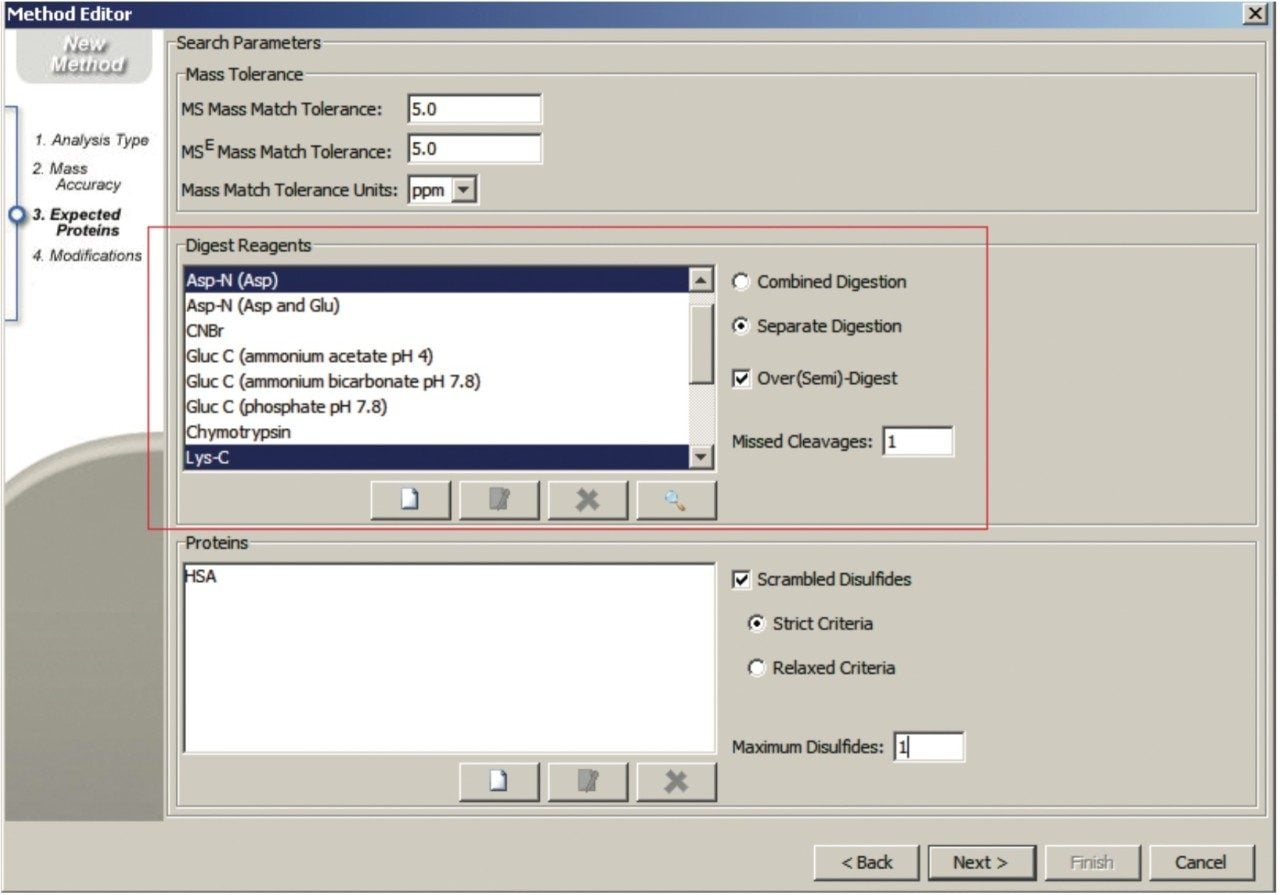
Combined multi-digest workflow
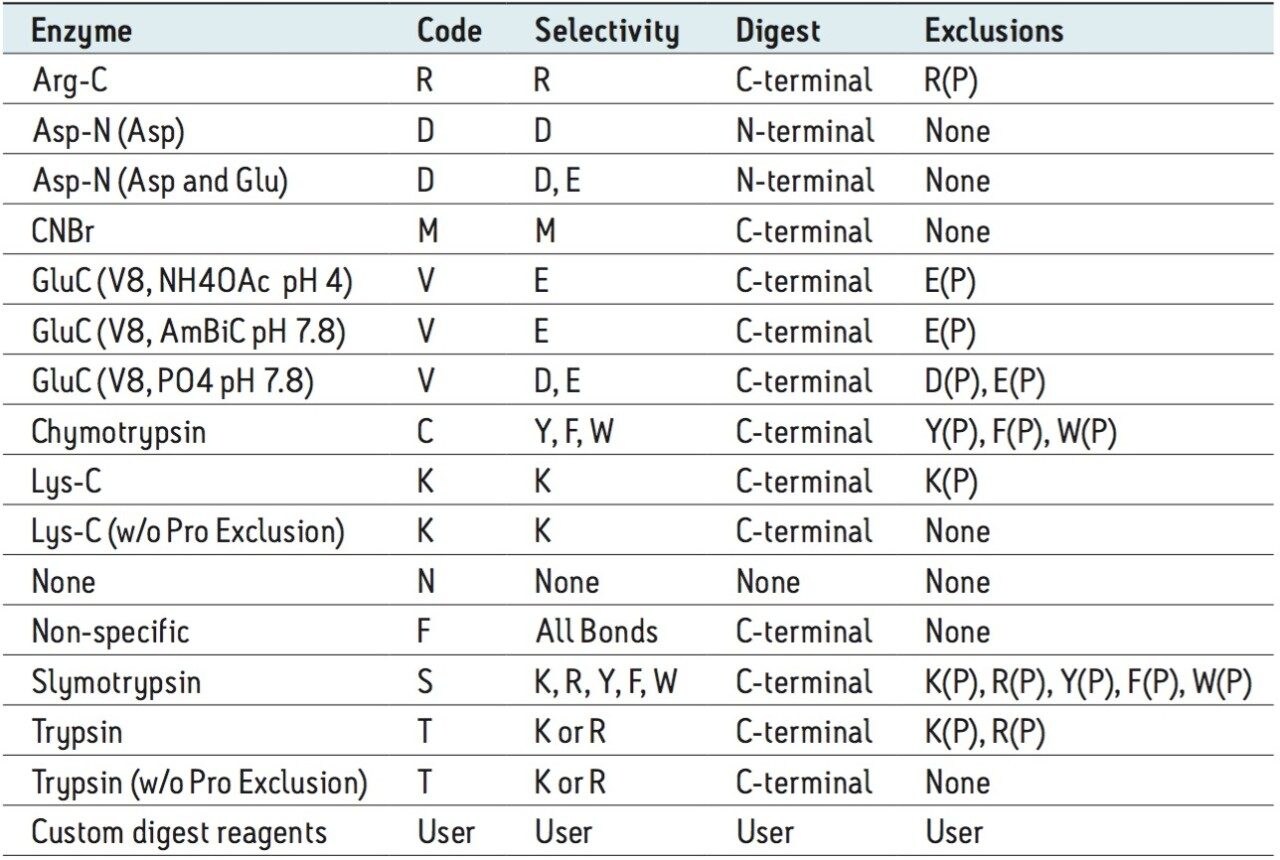
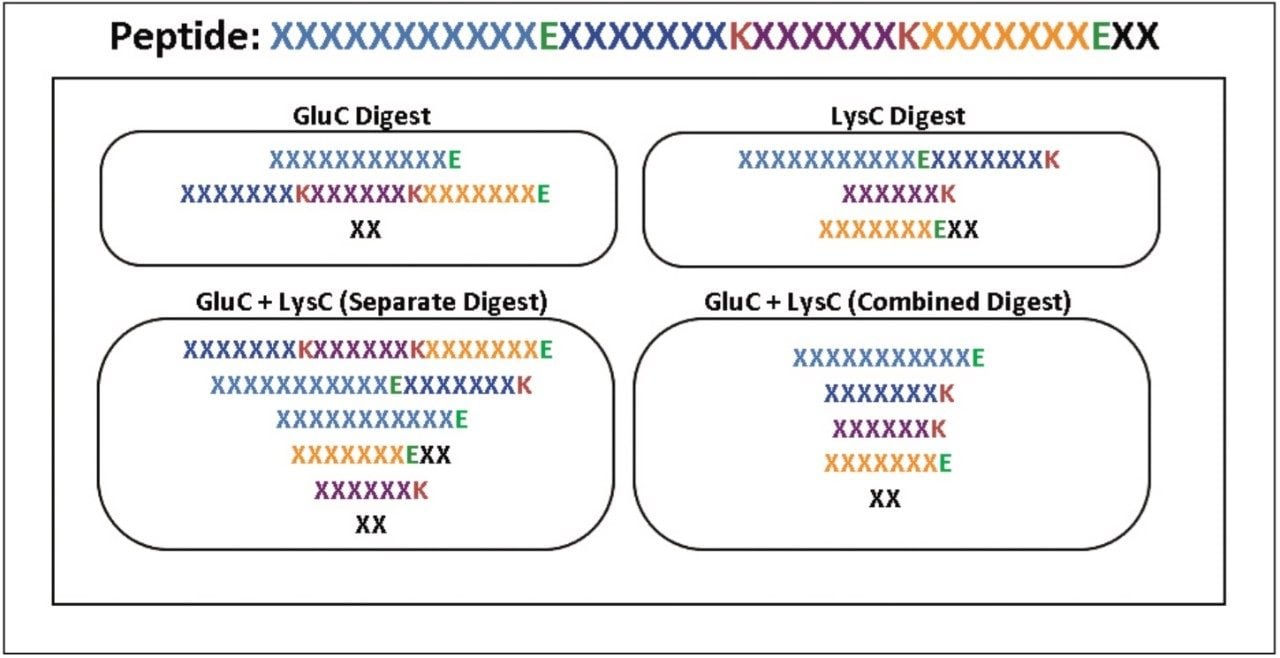
The combined workflow generates a set of peptides resulting from the combined specificities of all proteases used in the digestion process. Identified peptides are labeled using multi-letter peptide digest labels derived from the single enzyme digest designators (e.g., DT represents the combined product of AspN(D) and Trypsin (T)).
There are three common reasons to utilize the combined digest multi-enzyme workflow:
- To change the peptide digest population, altering chromatographic properties of the mixture. This will often introduce significant changes in retention time for multiple peptides, and can address peptide coelution, or improve chromatographic quality for one or more components.
- To reduce large peptides obtained from a single enzyme digest to more manageable-sized peptides for fragmentation studies. It can be difficult to generate fragmentation at all peptide bonds when sequencing larger peptides (> 25 AA). Reducing peptide size can often enhance fragment coverage for peptides of interest, and can be particular useful for producing smaller peptides where multiple modifications can be monitored independently.
- Changes to the terminal amino acid residues can significantly alter peptide fragmentation behavior and allow more confirmatory ions from sequence regions not favored for fragmentation of the larger peptide.
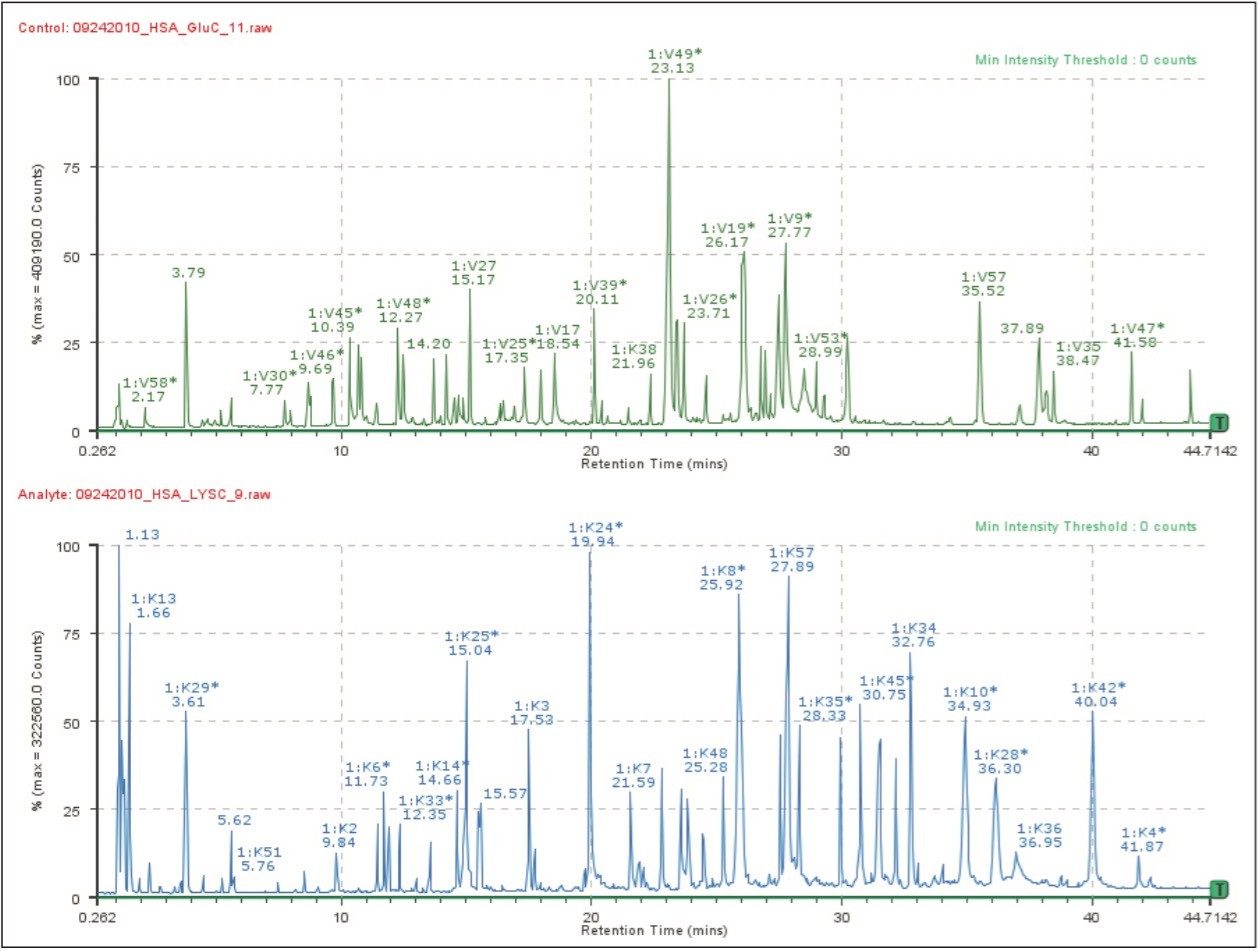
Separate multi-digest workflow
The separate workflow assumes that multiple independent digests were produced and that the enzymes were inactivated before the digests were mixed for analysis. BiopharmaLynx searches peptide mapping results for peptides predicted using the digest specificities of each enzyme. Identified peptides are labeled using the single-letter peptide digest nomenclature common to single enzyme digests (e.g., T for Trypsin; K for LysC; D for AspN)
There are three common reasons to utilize the separate digest multi-enzyme workflow:
- To obtain high sequence coverage with minimal method optimization for a given protein. This could prove useful for discovery scientists faced with evaluating large numbers of protein candidates, or during clone selection for a particular candidate.
- To obtain redundant protein coverage from a single peptide map. Modifications can be independently confirmed and quantitated using overlapping peptide sequences from each digest.
- To maximize sequence confirmation from peptide fragmentation using fragmentation selectivity differences between digested peptides covering a common sequence region.