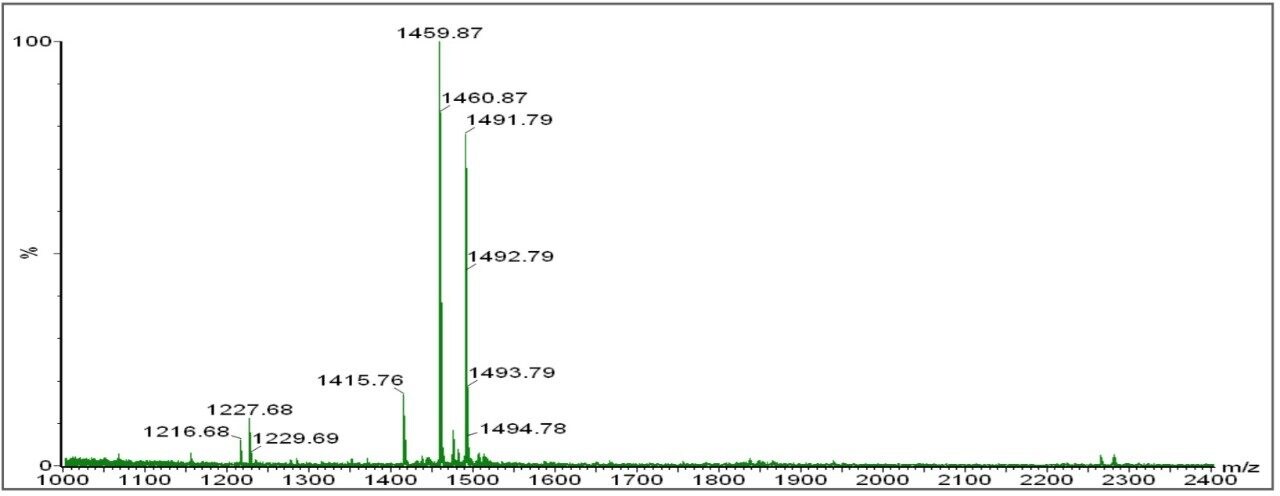
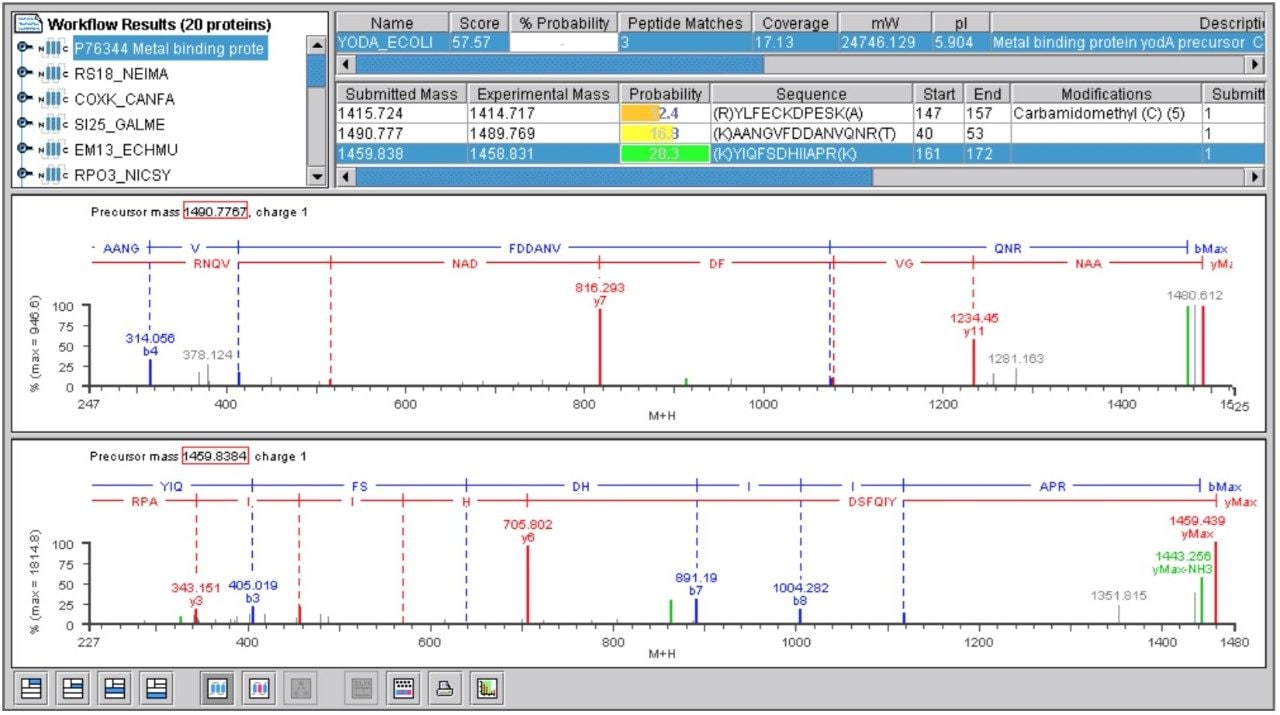
Databank Search Results
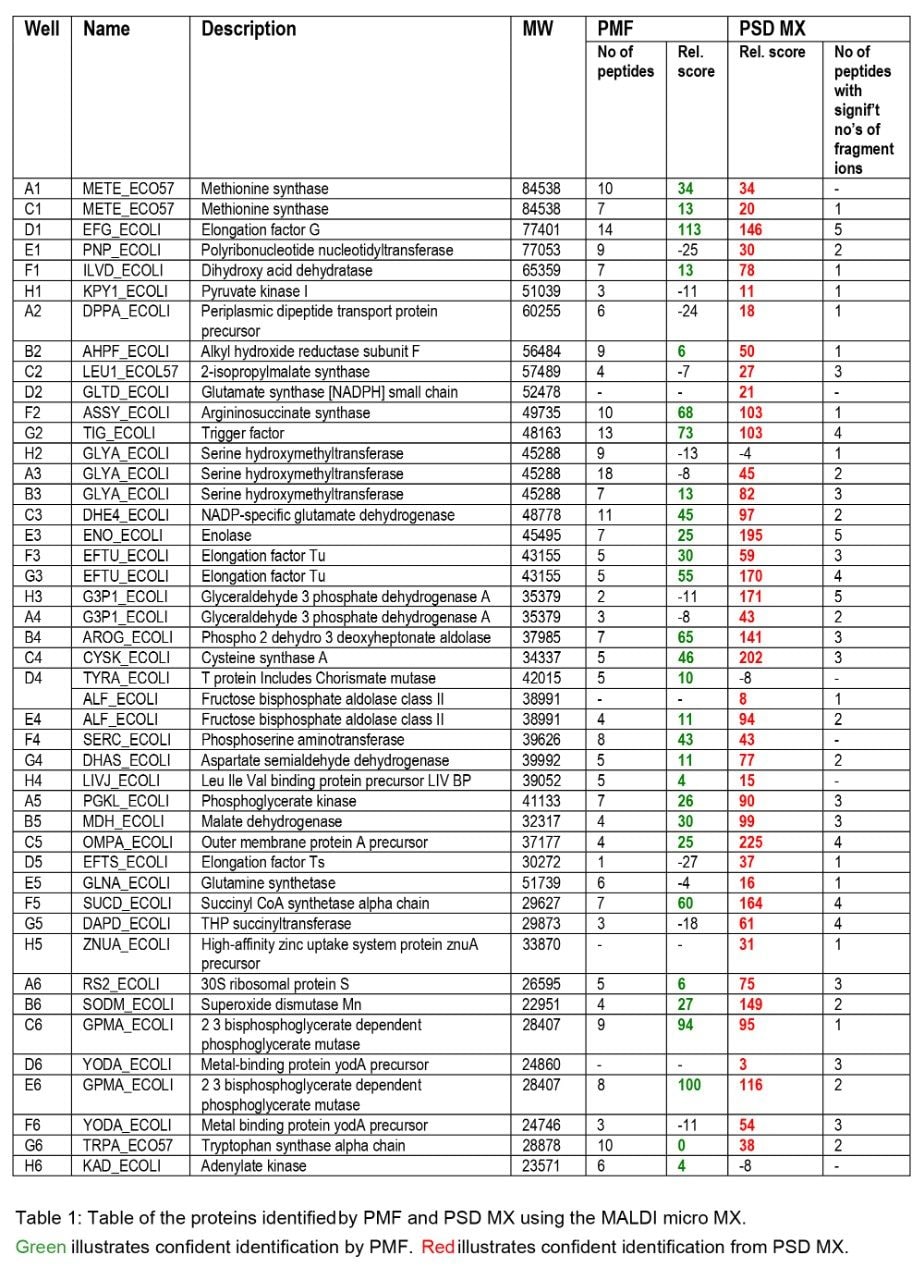
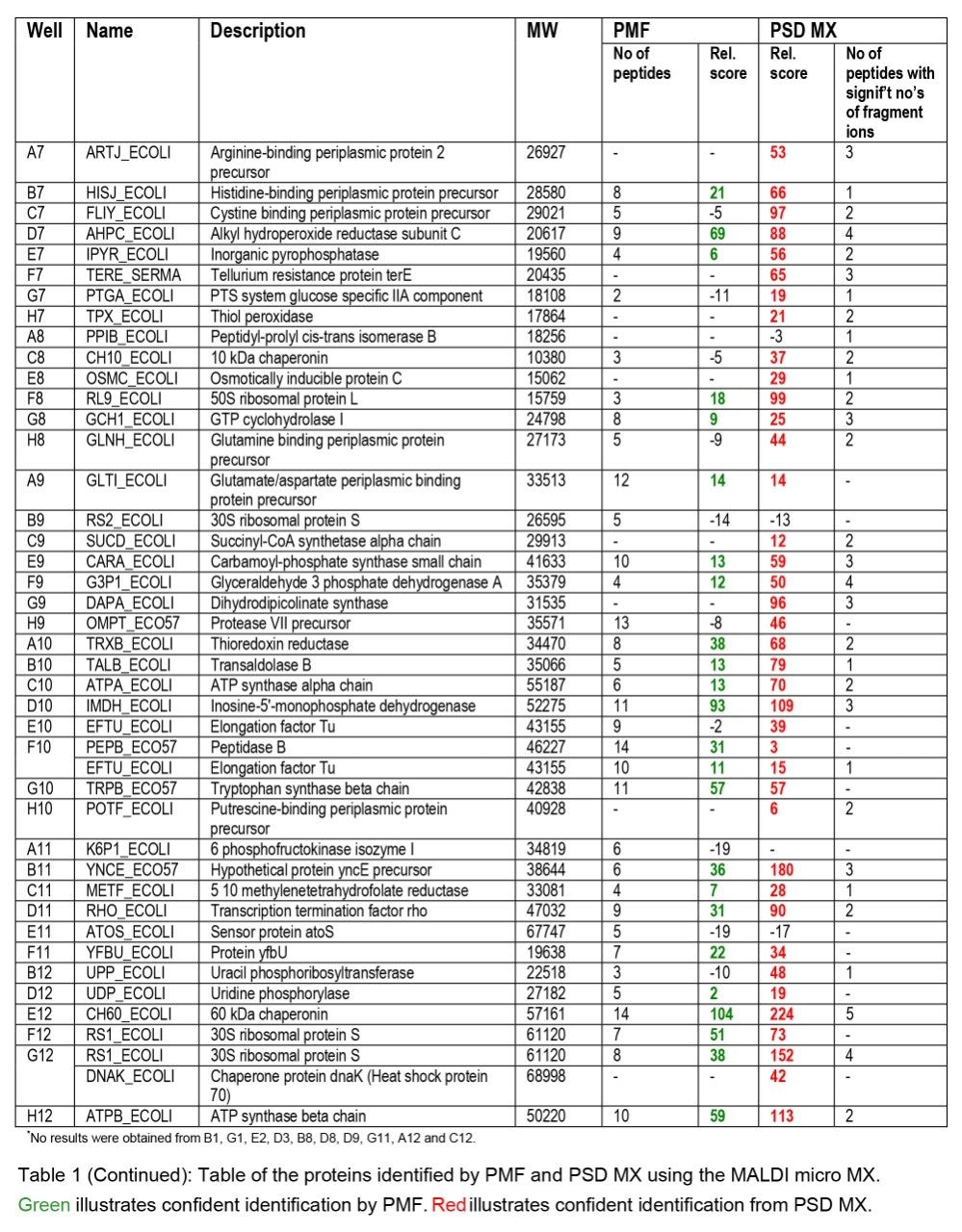
Databank search results obtained from the E. coli 2D gel samples are summarized in Table 1. Two different types of databank search were performed on the MS or PSD data obtained from each sample well. MS data were submitted for a peptide mass fingerprint search while PSD data were submitted using the fragment ion information. The search results from the fragment ion search were formatted as a “protein report”, described here as PSD MX. The two different types of score have a 95% confidence limit, which is governed by the search parameters used, the limit for PMF and PSD MX scores is a score of 64. To facilitate comparing different types of databank search/score a relative score was devised. The relative score was obtained by using the following equation:
Score relative= Score(indiv. Protein)- Score(95% confidence)
Therefore, the relative score can be calculated by subtracting the 95% confident protein identification level, (64 for both PMF and PSD MX) from the returned databank score for each individual protein.
In the case of tenuous or ambiguous protein identification a negative relative score will be produced, while positive relative scores suggest statistically significant, or unambiguous protein identification.
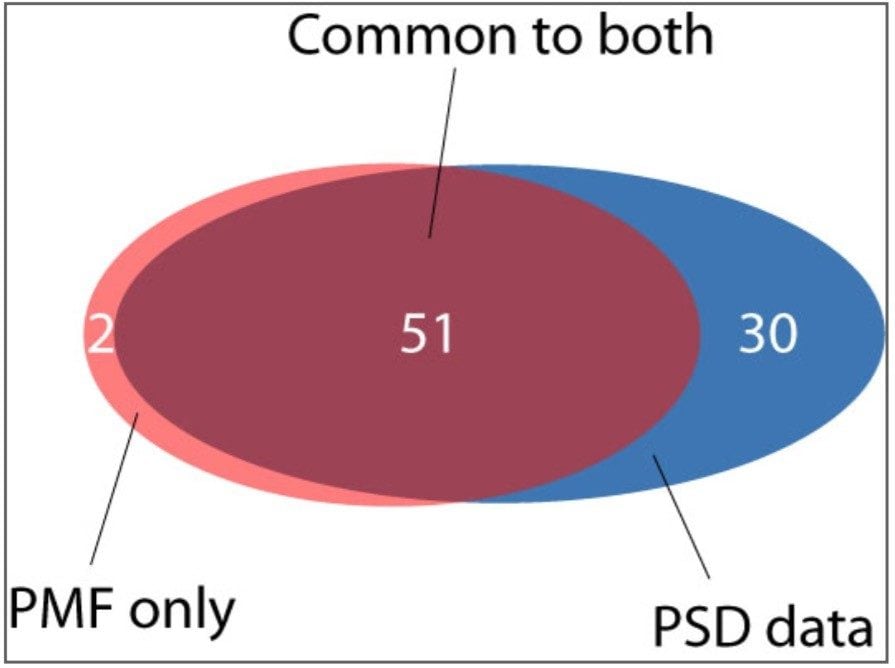
A closer examination of the resulting data indicates that using the traditional PMF approach 53 proteins were confidently identified from 96 sample wells (c.f. Table 1). It was possible to match two proteins to the MS data from well F10 using PMF. These search results represent successful protein identification from 54% of the samples. In our experience, this is a typical success rate for PMF protein identification. In contrast, databank searching of the PSD MX data, in combination with the PMF information, resulted in confident identification of 81 proteins, equivalent to a protein identification success rate of 82% from the samples analyzed. Of the 81 proteins identified, 30 proteins were only unambiguously identified when PSD MX data were considered. In addition, two proteins were found exclusively by using the PMF approach. A summary of the proteins identified by the different techniques used is illustrated graphically in Figure 1.