A Discovery Lipidomic Workflow Combining the Xevo™ MRT P10 with mzmine Data Processing Pipelines
Ansgar Korfa, Robin Schmida, Steffen Heuckerotha, Lee A. Gethingsb, Samantha Ferriesb, Nyasha Munjomab, David Heywoodb, Tomas Pluskala,c
amzio GmbH, Germany
bWaters Corporation, Wilmslow, United Kingdom
cIOCB, Prague, Czech Republic
Published on June 01, 2026
Abstract
A comprehensive lipidomic workflow requires both broad analyte coverage and high confidence structural annotation. In this study, liquid chromatography combined with high resolution mass spectrometry (LC-HRMS) data acquired using data dependent acquisition (DDA) and data independent acquisition (DIA) on the Xevo MRT P10 Multi-Reflecting Time-of-Flight (TOF) Mass Spectrometer were evaluated using an integrated mzmine based processing pipeline. DDA provided higher total lipid identifications and faster processing, while DIA improved coverage for selected sphingolipid subclasses. The combination of rule based annotation, multi criteria quality scoring, Equivalent Carbon Number (ECN) modelling, Kendrick Mass Defect (KMD) analysis, and Interactive Molecular Networking enabled confident lipid annotation and rapid identification of outliers or mis-assignments. This application note demonstrates how Waters MRT technology, paired with advanced open source data processing, supports deep and reliable lipidome characterization.
Benefits
- A seamless workflow for processing lipidomic high resolution MS data, providing broad lipid coverage and high-confidence structural annotation.
- DDA delivers higher total lipid identifications and faster processing, while DIA enhances coverage for specific sphingolipid subclasses.
- Comprehensive data provided by the Xevo MRT P10 Mass Spectrometer when combined with the advanced mzmine workflow—including rule‑based annotation, ECN modelling, KMD analysis, and Interactive Molecular Networking—produces reproducible, high‑quality MS/MS data suitable for confident and robust lipid identification.
Introduction
Lipidomics by LC-HRMS is challenged by the structural diversity of lipids and the complexity of MS/MS data. Acquisition strategy plays a critical role, whereby, DDA offers high quality MS/MS spectra but may miss low abundance or stochastically selected precursors. Alternatively, DIA can provide reproducible fragmentation across the mass range but produce more complex spectra requiring deconvolution.
The Xevo MRT P10 Mass Spectrometer platform provides high resolution, high mass accuracy MS and MS/MS data suitable for both acquisition modes. To fully exploit these datasets, an mzmine workflow was applied that incorporated feature detection and alignment, rule based lipid annotation with quality scoring, ECN retention time modelling and KMD analysis for homologous series. Additional features such as Interactive Molecular Networking for annotation propagation, allows for visual organization of chemical relationships, while the new Lipid Annotation QC Dashboard, allows a rapid review of datasets to ensure consistent and high-quality analyses. This application note compares lipidome coverage and annotation confidence between DDA and DIA using this unified workflow.
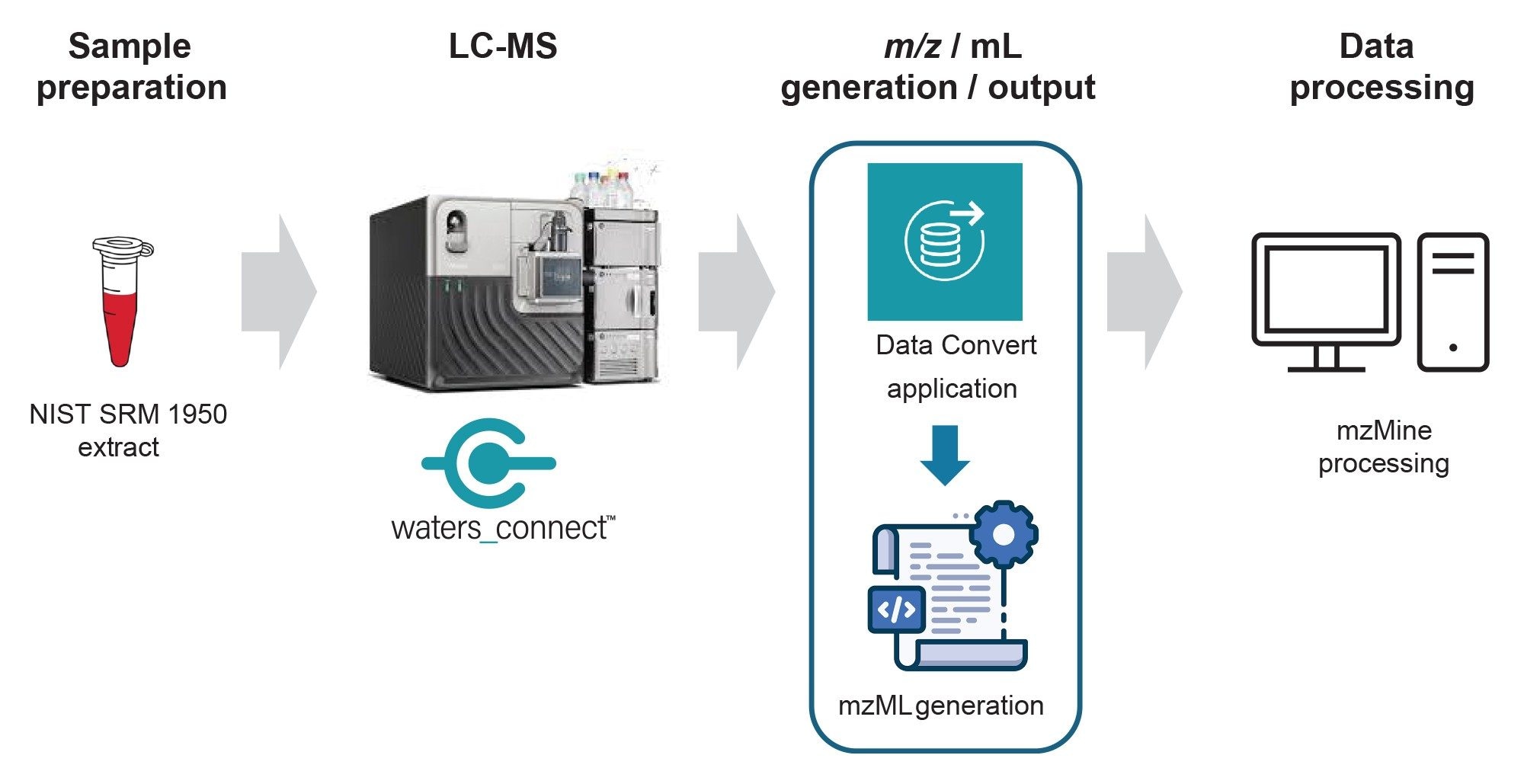
Experimental
Sample Preparation for LC-MS
Lipids were extracted from the human plasma standard, NIST SRM 1950 (Sigma-Aldrich, Poole, UK). Extraction was conducted using the Bligh dyer method.1 Extracts were resuspended with methanol and injected as technical replicates (n=3). Inject volumes consisted of 1 and 2 µL for positive (ESI+) and negative (ESI-) ion respectively.
LC-MS Data Acquisition (DDA)
LC-MS data were collected using an ACQUITY™ Premier UPLC System coupled to a Xevo MRT P10 Mass Spectrometer. The reversed-phase (RP) chromatographic separation consisted of a 12 minute method (injection to injection) using an ACQUITY Premier CSH C18, 1.7 µm, 2.1 x 100 mm analytical column (p/n: 186009461). The LC solvents comprised of mobile phase A (600:390:10; ACN: water:ammonium formate (10 mM) (v/v), 0.1% formic acid) and mobile phase B (900:90:10; IPA: ACN:ammonium formate (10 mM) (v/v), 0.1% formic acid). A 10 minute gradient (12 minute turnaround) was implemented, starting with a solvent composition of 50% mobile phase B. Over 7.0 minutes, mobile phase B was ramped to 80% before reaching 99% at 10 minutes. The solvent composition was held at 99% (mobile phase B) for 1.0 minute and re-equilibrated to initial conditions by 2 minutes, ready for the subsequent injection. Samples were injected as technical triplicates for both ionization modes.
The Xevo MRT P10 Mass Spectrometer was configured with the source settings as outlined in Table 1. MS data were collected using DDA in both positive and negative ion modes. DDA methods consisted of scan rates of 10 Hz (MS) and 20 Hz (MS/MS). A top 30 (ESI+) and top 20 (ESI-) setting were applied with a dynamic exclude of 5 ppm and a 2 second retention time tolerance. The quadrupole isolation window was set to 1 Da for all acquisitions.
|
Parameter |
Value |
|
Capillary voltage (kV): |
2.5/1.2 (ESI+/ESI-) |
|
Sample cone (V): |
30 |
|
Desolvation gas (L/min): |
700 |
|
Cone gas (L/min): |
20 |
|
Desolvation temperature (°C): |
500 |
|
Source temperature (°C): |
120 |
Table 1. Xevo MRT P10 Mass Spectrometer source settings.
LC-MS Data Acquisition (DIA)
DIA data were collected using the Xevo MRT P10 Mass Spectrometer using the same LC conditions and MS source settings as described for the DDA experiments. The DIA method (MSE) settings are detailed in Table 2.
|
Parameter |
Value |
|
Scan rate (Hz): |
20 (10 MS1; 10 MS2) |
|
Collision energy (eV): |
25–55 (linear ramp) |
|
Mass range: |
50–1200 m/z |
Table 2. DIA (MSE) settings.
Data Processing
Data were exported as .mzML automatically during data acquisition via the Data Convert Application within waters_connect™ for Data Readiness Software. The acquired data were processed using mzmine. Data were peak picked and identifications provided by searching against a reference lipid database.
Results and Discussion
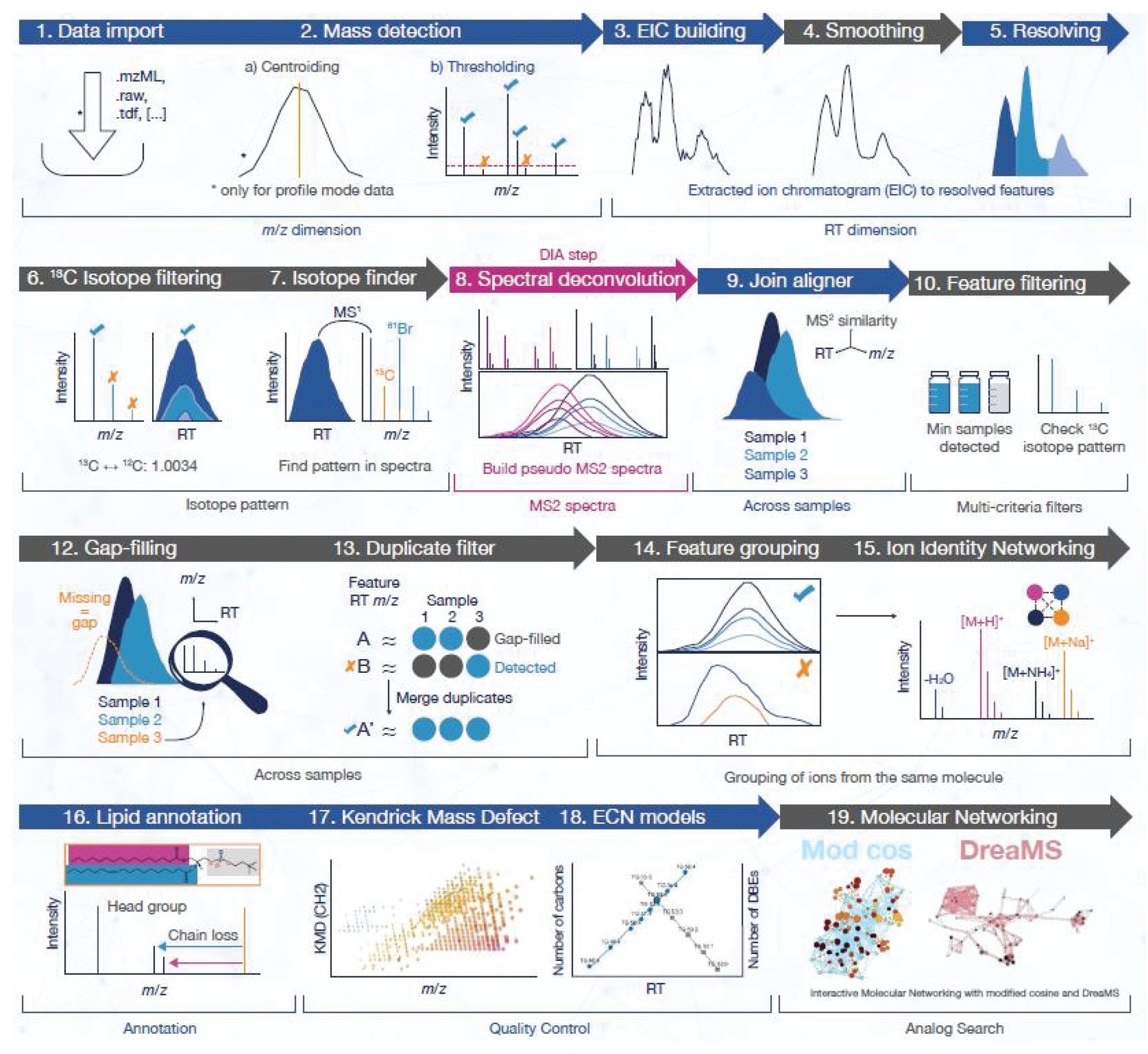
Data relating to the lipid extracts from the NIST SRM 1950 sample were collected and processed as shown in Figure 1. Individual steps related to the processing within mzmine are outlined in Figure 2. In short, the data undergo detection, smoothing, and isotope filtering. For DIA related data, there is an additional step (shown as step 8; Figure 2), whereby pseudo MS2 spectra are built as part of spectral deconvolution. The latter stages of the processing workflow include gap-filling/duplicate filter, for those cases where there may be missing data values between samples/replicates. In order to identify features based on MS1 which may be linked (i.e., same species but present as multiple adducts, related through in-source fragments etc), uses Ion Identity Networking (IIN). The compound identification stage also uses a dedicated lipid annotation tool prior to generating KMD plots and ECN models to identify and correlate lipid categories and classes.
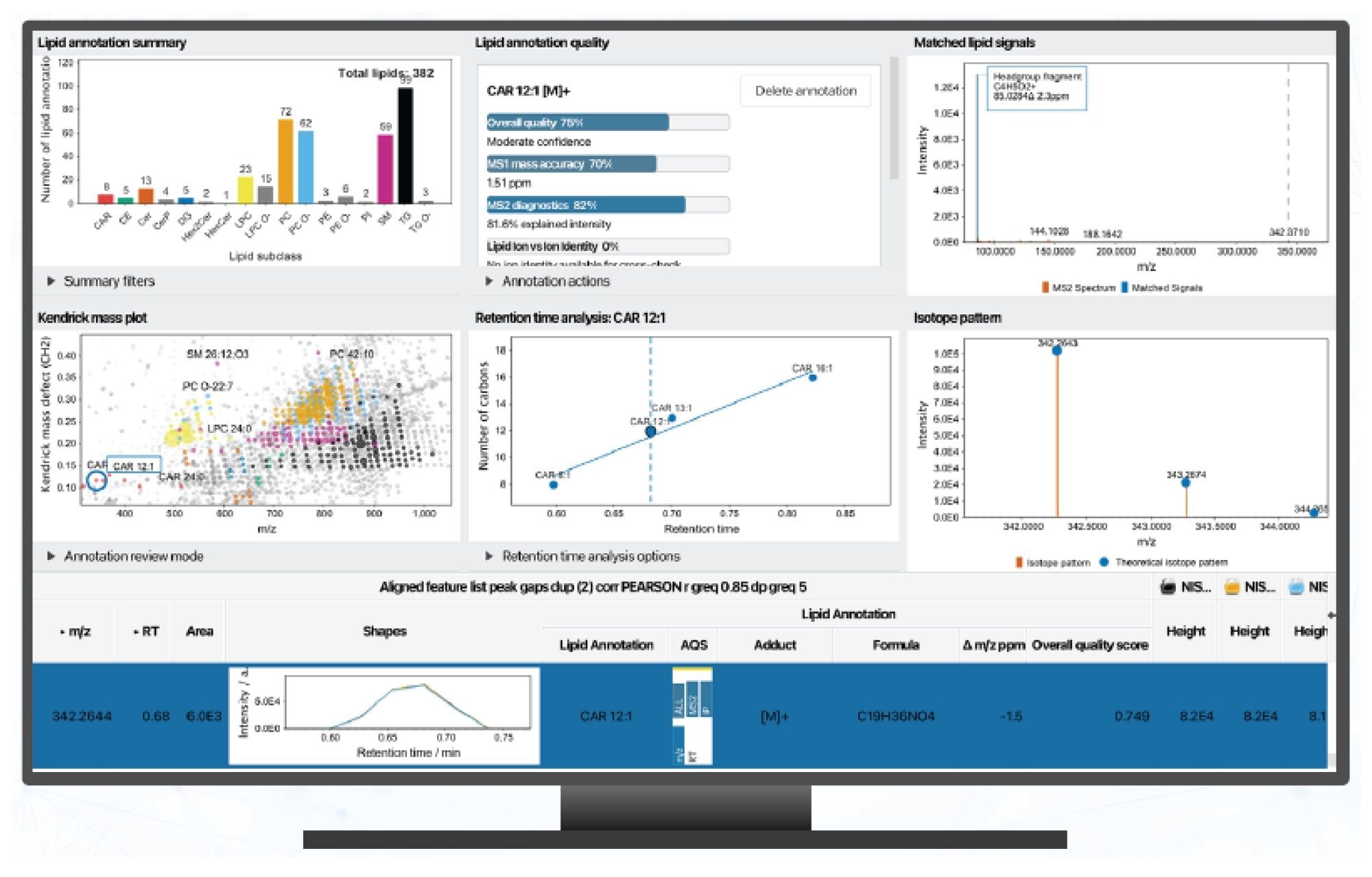
Reviewing the processed data highlighted that both acquisition modes produced highly reproducible MS/MS spectra, enabling confident rule based lipid annotation. The ability to review data via the Lipid Annotation QC Dashboard, allows for a quality check of annotations in addition to identifying potential false positives/negatives using a variety of methods such as RT distributions and comparing isotope patterns (Figure 3).
Other features of the dashboard, include MS/MS diagnostic fragment evaluation, duplicate annotation detection, KMD plots to visualize homologous series, and ECN modelling to verify class specific retention behavior. The results for the data shown in Figure 3, revealed consistent RT behavior across all the lipid subclasses (based on ECN modelling). The KMD plot provided visualization of homologous lipid series and differences in data completeness between the DDA and DIA datasets.
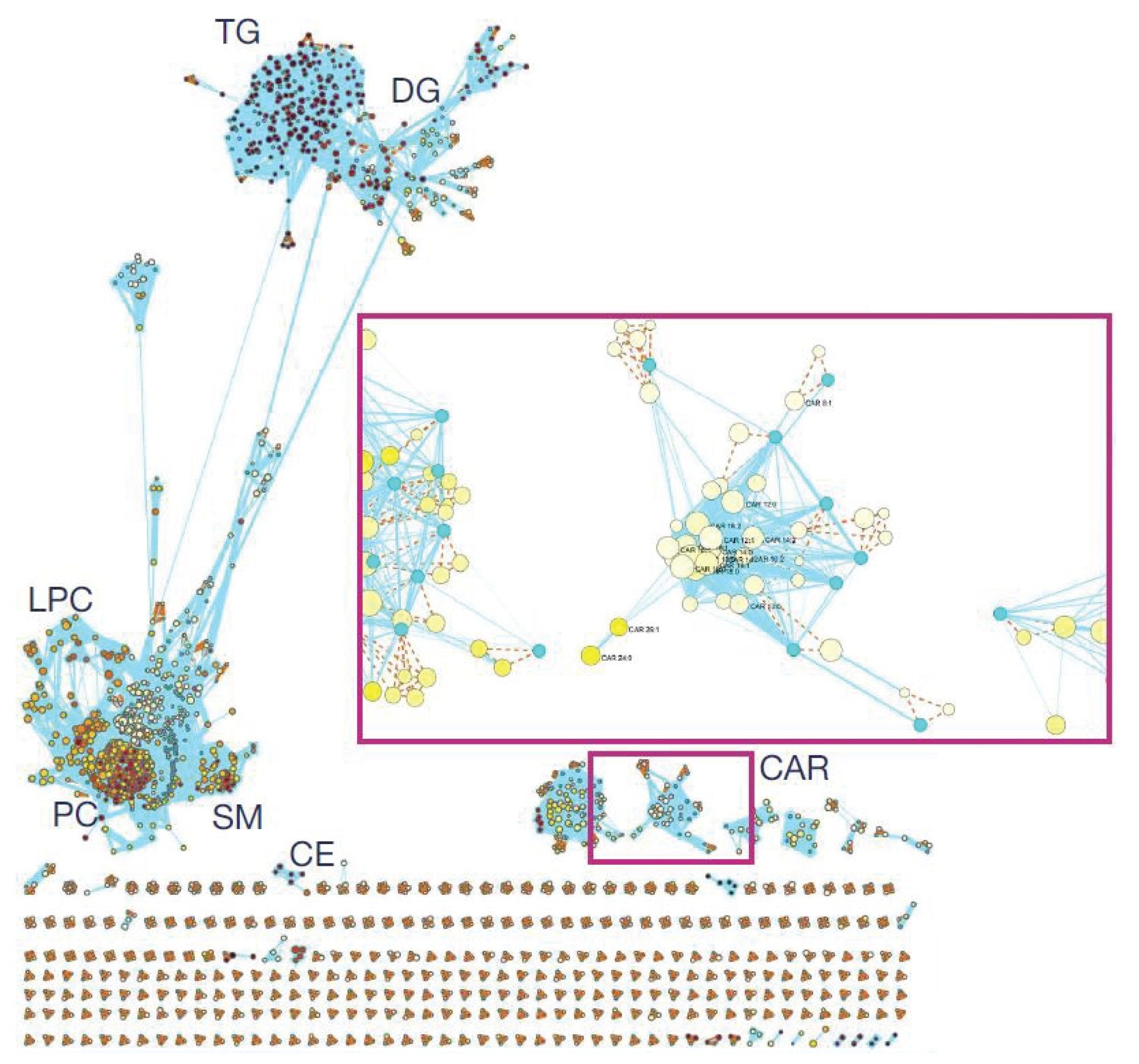
An additional feature of mzmine, which is particularly useful, is Interactive Molecular Networking. The principle of which is to cluster lipid species by lipid class, enabling the discovery of missing or incomplete annotations by linking unknown features to annotated lipid families based on MS2 similarity (Figure 4).
Comparing the DDA and DIA datasets resulted in DDA providing more total lipid identifications with the added benefit of shorter processing times. The coverage of lipid classes also varied between both acquisition modes, with a higher coverage of phospholipids and headgroup-identified sphingolipids from DDA-based acquisitions. Interestingly, selected sphingolipid subclasses such as HexCer had greater coverage with DIA, most likely as a result of its comprehensive fragmentation (Figure 5). Comparing the fragmentation quality between both modes of acquisition, showed that it was consistent across both modes of acquisition and therefore supporting robust annotation. These trends align with expected behavior: DDA excels in targeted MS/MS quality, while DIA ensures systematic sampling of low abundance species.
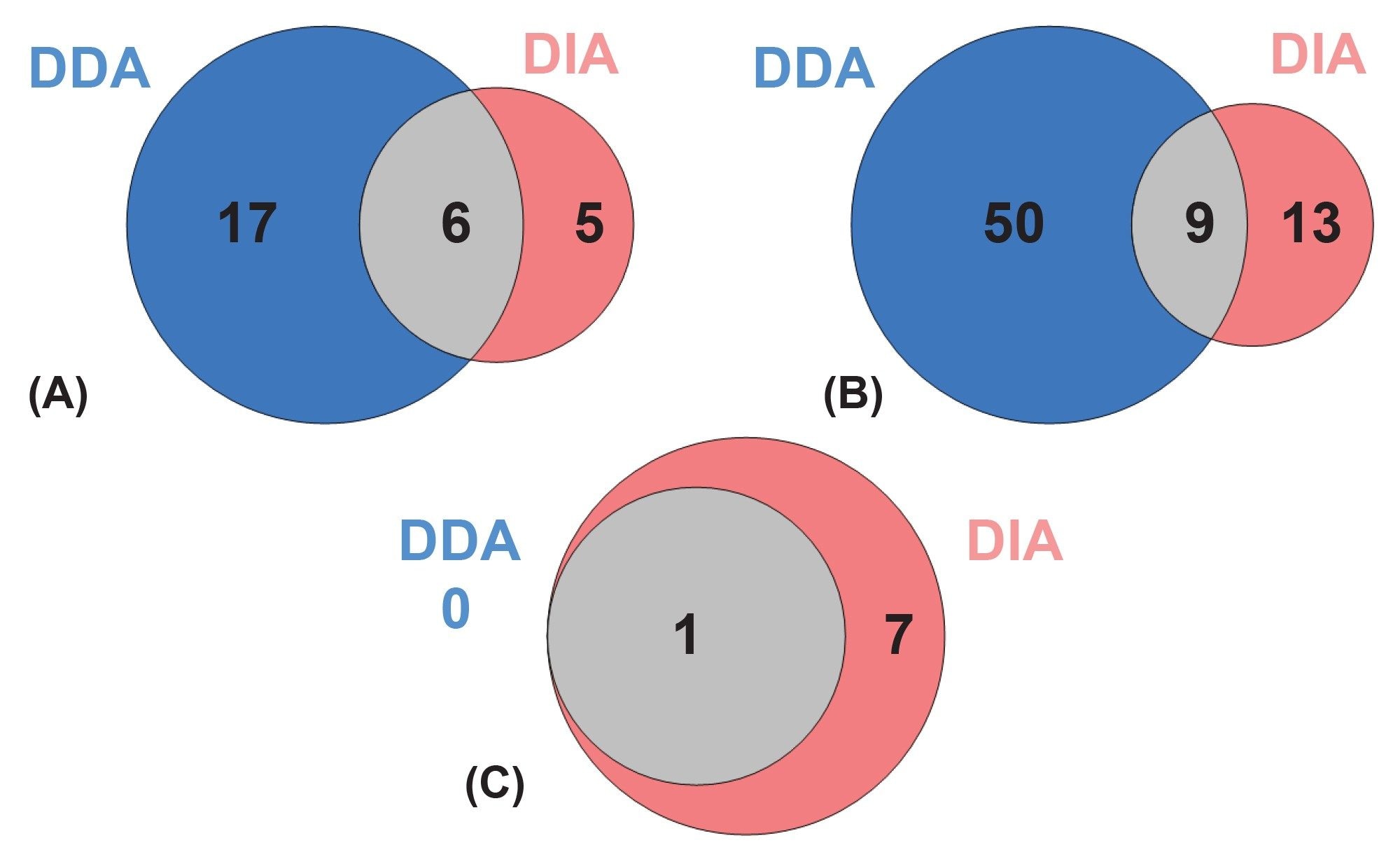
Conclusion
This study demonstrates that both DDA and DIA on the Xevo MRT P10 platform generates high quality fragmentation suitable for confident lipid annotation. DDA delivered higher total lipid identifications and faster processing, whilst DIA improved coverage for specific sphingolipid subclasses. The integrated mzmine workflow—combining rule based annotation, ECN modelling, KMD analysis, and Interactive Molecular Networking—provided a transparent, reproducible, and high confidence lipidomic pipeline. The Lipid Annotation QC Dashboard enabled rapid review and correction of potential mis annotations. Together, these tools support deep lipidome exploration with robust annotation reliability.
References
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology., 1959, 37 (8), 911–17.
720009365, June 2026