乳制品中氨基酸的UPLC™-UV分析 - 在ACQUITY™ Premier系统上实施国际标准
摘要
本研究采用反相超高效液相色谱法对乳制品中的氨基酸进行分析,使用配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统,并严格按照AOAC国际官方分析方法2018.06(AOAC方法2018.06)进行实验通过对色谱条件和操作流程的优化,包括流动相A的制备、进样模式与参数设置、衍生化配方以及在线过滤器的使用,实现了更可靠、高效的氨基酸分离。通过这些改进,两种标准品和一系列代表性乳制品均实现了可靠的氨基酸分离,并获得了优异的线性、灵敏度、精度、峰形以及基线分离。本应用纪要展示了在配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统上成功应用AOAC方法(2018.06)的实例。
优势
- AOAC方法2018.06为乳制品中的氨基酸分析提供了更快速、更全面的解决方案
- 配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统为采用AOAC方法2018.06对氨基酸进行高效分离提供了可靠的解决方案
- AccQ∙Tag™ Ultra衍生化试剂盒和AccQ∙Tag Ultra化学品试剂盒简化了AOAC方法2018.06的实施流程
简介
氨基酸(AA)是人类和动物的生长和维持健康的必需营养素。氨基酸总含量及氨基酸谱具有重要的营养学价值。AA谱也是某些食品的特征指标,可用于甄别可能的掺伪行为。传统的AA分析方法(例如AOAC方法994.12和AOAC方法985.28)需经历过甲酸氧化、酸水解、离子交换色谱分离及茚三酮柱后衍生等步骤,其中耗时漫长的过甲酸氧化过程会导致酪氨酸结构破坏,而需要单独分析。AOAC方法2018.06提供了一种更快速、更全面的AA分析方法1。 在该方法中,样品经过酸水解,然后用6-氨基喹啉-N-羟基琥珀酰亚胺基甲酸酯(AQC)进行柱前衍生化,接着采用反相超高效液相色谱(RP-UPLC)分离与紫外/可见光(UV/Vis)检测联用,不仅省去了过夜过甲酸氧化步骤,还可实现酪氨酸与其他氨基酸的同步分析。AOAC方法2018.06已被国际标准化组织(ISO) 4214:20222、国际乳品联合会(IDF) 254:20222,以及美国国际谷物化学家学会(AACC) 07–50.01等机构认证为官方标准3。
沃特世的氨基酸分析解决方案4,5在柱前衍生化处理(采用AQC)和RP-UPLC方面采用了相似的方法。唯一的区别是,在RP-UPLC中,AOAC方法2018.06使用了更长的色谱柱。因此,沃特世的多款氨基酸分析产品已被应用于AOAC方法2018.06中。这些产品包括ACQUITY UPLC BEH™ C18(1.7 µm,2.1 × 150 mm,P/N:186002353)、AccQ∙Tag™ Ultra衍生化试剂盒(186003836)、AccQ∙Tag Ultra洗脱溶液A浓缩液(P/N:186003838)和AccQ∙Tag Ultra洗脱溶液B(P/N:186003839)。除了色谱柱和消耗品以外,参与多实验室研究的多个实验室还成功使用了多款Waters ACQUITY系统,包括ACQUITY UPLC系统、ACQUITY UPLC H-Class系统和ACQUITY UPLC I-Class系统,并获得了优异的结果,促成了AOAC方法2018.06的批准6。鉴于AA分离易受LC仪器硬件和设计的影响,我们评估了配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统,以实施AOAC方法2018.06。研究中特别关注了实现可靠、高效AA分离的色谱条件。
实验
本研究遵循了AOAC方法2018.06中规定的实验细节,并进行了少量调整。以下简要介绍了关键步骤和条件。更多详细信息可参见AOAC方法2018.06。
AA校准标准品
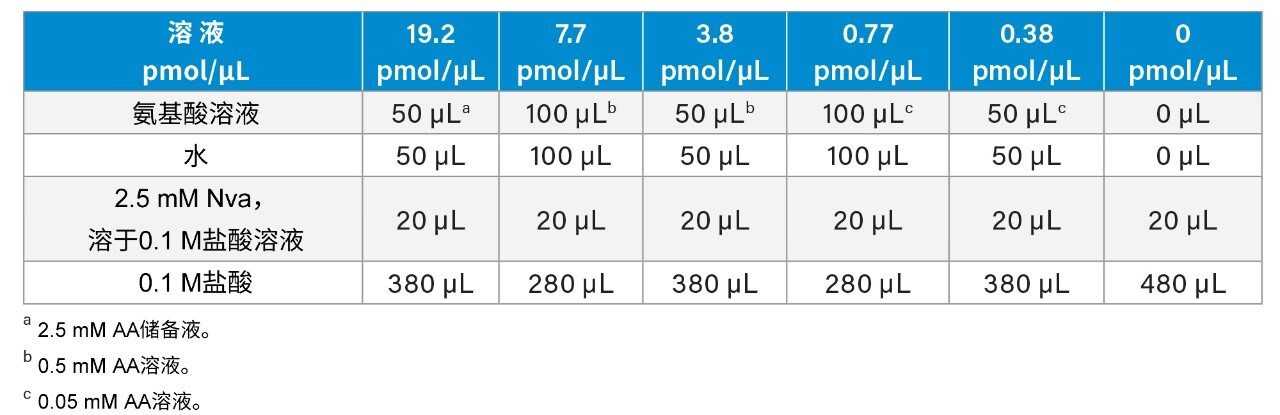
2.5 mM AA储备液:AA储备液(沃特世P/N:WAT088122)包含以下每种氨基酸(除L-胱氨酸1.25 mM外,其余浓度为2.5 mM,溶于0.1 M盐酸):L-丙氨酸(Ala)、L-精氨酸(Arg)、L-天冬氨酸(Asp)、L-胱氨酸(Cys)、L-谷氨酸(Glu)、L-甘氨酸(Gly)、L-组氨酸(His)、L-异亮氨酸(Ile)、L-亮氨酸(Leu)、L-赖氨酸(Lys)、L-甲硫氨酸(Met)、L-苯丙氨酸(Phe)、L-脯氨酸(Pro)、L-丝氨酸(Ser)、L-苏氨酸(Thr)、L-酪氨酸(Tyr)和L-缬氨酸(Val)。使用0.1 M盐酸连续稀释,从AA储备液中制备了0.5 mM和0.05 mM的AA溶液。根据表1制备浓度为0~19.2 µM (pmol/µL)的AA校准溶液和7.7 µM的正缬氨酸(Nva)(均为衍生化后的最终浓度)溶液。使用Nva作为内标(I.S.)。
注:尽管该溶液中包含胱氨酸,但未用于定量分析,且单独制备。
此外,沃特世食品和饲料AA标准溶液(沃特世P/N:186009299)也可用于AA校准标准品。该溶液包含17种AA(0.5 mM,与AA储备液中的AA相同),另含0.5 mM的以下AA:牛磺酸(Tau)、α-氨基丁酸(AABA)、甲硫氨酸砜(MetSO2)和磺基丙氨酸(Cya)。
胱氨酸校准标准品
称取240 mg胱氨酸(精确至0.01 mg),加入100 mL容量瓶中,用0.05 M NaOH定容,制得胱氨酸储备液(10 mM)。用0.05 M NaOH稀释胱氨酸储备液,制得胱氨酸溶液(1 mM)。10 mM胱氨酸储备液可在–20 ℃下保存至多3个月。每次分析中的1 mM胱氨酸溶液需现用现配。
分别移取0、10、20、50、100和200 µL的10 mM胱氨酸储备液至10 mL样品瓶中,用水定容至1100 µL,制备胱氨酸校准标准品。向每个样品瓶中加入600 µL DDP(3,3'-二硫代二丙酸)溶液(0.2 M NaOH中含1% DDP)、600 µL 0.2 M盐酸、200 µL 10mM Nva储备液,和2500 µL苯酚-盐酸溶液(12 M盐酸中含0.1%苯酚)。衍生化后这些标准溶液的最终浓度分别为0、0.38、0.77、1.9、3.8和7.7 µM (pmol/µL)。用氮气流喷射样品瓶至少5 s以置换氧气。旋紧样品瓶的螺口盖,在涡旋混合器中混匀。注:苯酚–盐酸溶液必须在通风橱内添加。
样品前处理
首先将婴儿配方奶粉和乳粉样品复溶,取2.5 g粉末加入40 g水中,充分混合。称取800±20 mg复溶样品,放入10 mL带有螺口盖的玻璃容器或样品瓶中。(样品质量精确至0.01 mg)向各容器或样品瓶中加入600 µL DDP溶液(0.2 M NaOH中含1% DDP)、600 µL 0.2 M盐酸、500 µL 10 mM Nva储备液和2500 µL苯酚–盐酸溶液(12 M盐酸中含0.1%苯酚)。
酸水解(仅适用于样品和胱氨酸标准品,不适用于AA标准品)
将装有样品溶液或胱氨酸标准曲线溶液的容器或样品瓶置于110 ± 2 °C的恒温箱中,加热24 ± 0.5 h。
或者,使用微波水解装置(Discover Prep,CEM公司,北卡罗来纳州)水解样品。使用35 mL硼硅酸盐玻璃压力容器(额定压力高达300 psi)(CEM P/N: 909036)和硅胶盖(CEM P/N: 909350)。使用涂覆有Teflon涂层的微型搅拌棒(CEM P/N: 162810)进行搅拌。酸水解步骤参数如下:动态控制,最大功率设置为300 W;温度设定为160 °C,持续20 min;压力限值设置为300 psi;搅拌级别设置为“高”;冷却过程中吹入氮气。
中和与稀释(样品和胱氨酸标准品)
酸水解完成后,等待水解产物冷却。然后,取每种水解产物0.2 mL,转移至1.5 mL微量离心管中,加入0.2 mL 6 M NaOH和1.6 mL 0.1 M盐酸。将混合物充分混匀,然后使用0.45 µm PVDF膜式过滤器将混合液过滤到另一个1.5 mL微量离心管中。
衍生化处理(样品、胱氨酸标准品和AA-标准品)
参照AOAC方法2018.06或AccQ∙Tag Ultra衍生化试剂盒7(沃特世P/N:186003836)维护和使用手册中的步骤进行AccQ∙Tag Ultra衍生化试剂配制,并对校准标准溶液、中和后的样品溶液或中和转化后的胱氨酸标准溶液实施衍生化(注:维护和使用手册中的步骤与AOAC方法2018.06中的衍生化步骤一致)。实际操作采用以下衍生化配方(体积比)替代手册中的原始配方:100 µL AccQ∙Tag Ultra硼酸盐缓冲液[AccQ∙Tag Ultra衍生化试剂盒中的试剂1]、10 µL校准溶液/中和样品溶液/中和转化后的胱氨酸标准溶液,以及20 µL复溶的AccQ∙Tag Ultra试剂(注:此优化方案有助于改善色谱峰形)。
UHPLC分离条件
|
UHPLC系统: |
ACQUITY Premier系统由二元溶剂管理器、固定定量环样品管理器、带主动预加热器的柱温箱(CH-A),以及光电二极管阵列(PDA) eλ检测器组成 |
|
UV/Vis波长: |
260 nm |
|
连接检测器的入口管路: |
0.004 in内径,10.5 in管路组件(沃特世P/N:430001784) |
|
软件: |
Empower™ 3色谱数据系统 |
|
色谱柱: |
ACQUITY UPLC BEH C18色谱柱, 1.7 µm, 2.1 mm × 150 mm(沃特世P/N:186002353) AccQ∙Tag Ultra C18色谱柱, 1.7 µm, 2.1 mm × 150 mm(沃特世,P/N:186009954) (注:AccQ∙Tag Ultra C18色谱柱是专为氨基酸分析定制的ACQUITY UPLC BEH C18色谱柱) |
|
色谱柱在线过滤器: |
ACQUITY 0.2 µm色谱柱在线过滤器(沃特世P/N:205000343) |
|
采集速率: |
10 Hz |
|
柱温: |
50 °C |
|
样品温度: |
20 °C |
|
样品定量环大小: |
1 µL |
|
进样体积: |
1 µL |
|
进样模式: |
带针溢出的部分定量环(PLNO),6倍溢出体积 |
|
流动相A: |
AccQ∙Tag Ultra洗脱溶液A浓缩液(沃特世P/N:186003838)与水的混合溶液 (1:6 (v/v)) (注:使用100 mL A型容量瓶移取100 mL AccQ∙Tag Ultra洗脱溶液A浓缩液,并与600 mL水混合。) |
|
流动相B: |
AccQ∙Tag洗脱溶液B(沃特世P/N:186003839) |
|
弱洗针液: |
95:5 (v/v)水:乙腈 |
|
强洗针液: |
5:95 (v/v)水:乙腈 |
|
密封清洗溶剂: |
50:50 (v/v)水:乙腈 |
|
流速: |
0.4 mL/min |
|
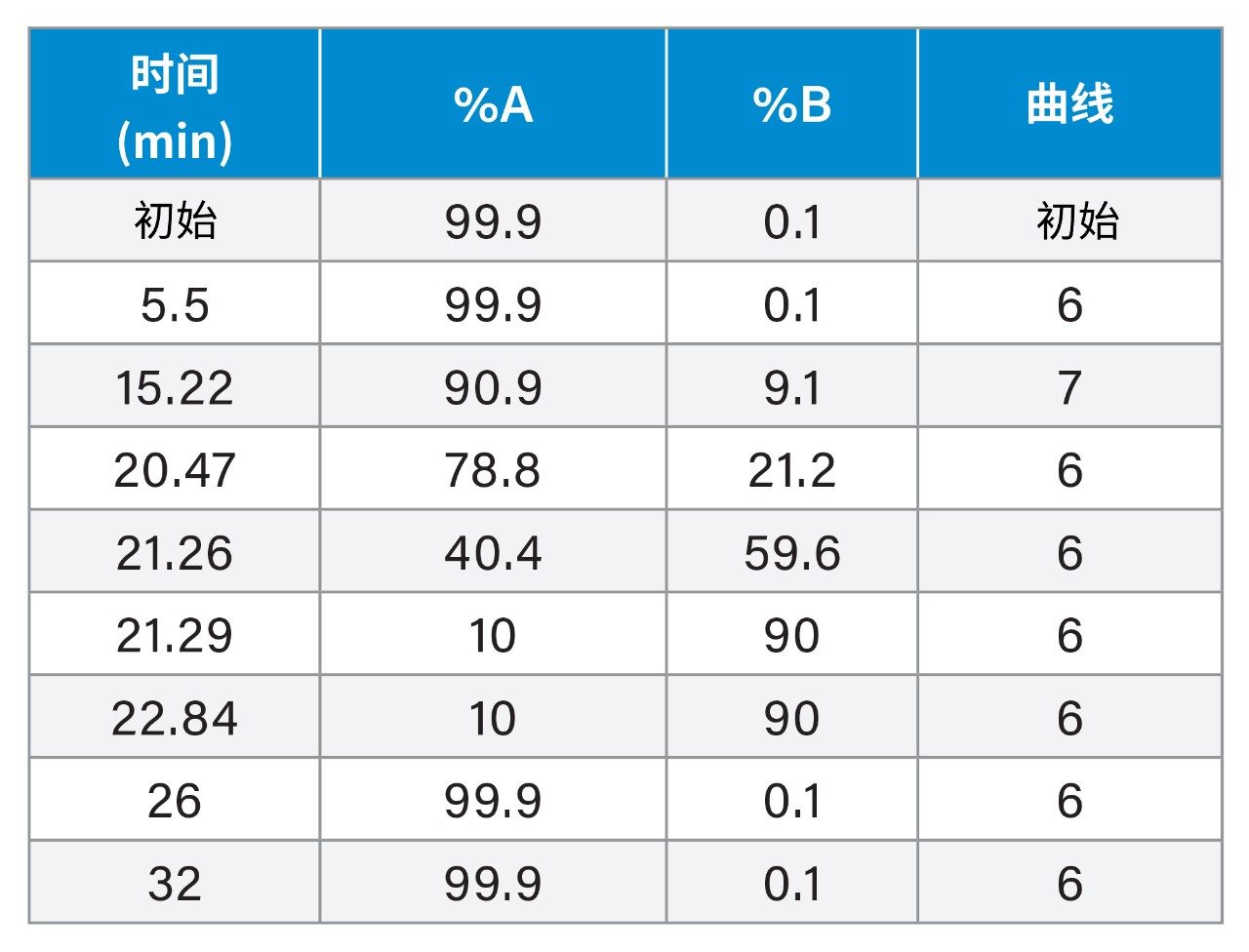
梯度洗脱: |
(请参阅下面的梯度表) |
梯度表
计算
计算详情请参阅AOAC方法2018.06。
结果与讨论
分析流程概述
根据AOAC方法2018.06,蛋白质在含有苯酚和DDP(3,3'-二硫代二丙酸)的6 M盐酸环境中进行水解。实验采用Nva作为内标。胱氨酸和半胱氨酸通过DDP转化为S-2-羧乙基硫代半胱氨酸(XCys)8。 水解中和后,AA和XCys经AQC衍生化处理,衍生化AA和XCys最终通过RP-UPLC进行分离,并在UV/Vis 260 nm下进行检测。在酸水解过程中,Gln和Asn分别转化为Glu和Asp。因此,Glu测定值为Glu和Gln的总和,Asp测定值为Asp和Asn的总和。XCys测定值则为半胱氨酸和胱氨酸的总和。色氨酸在酸水解条件下会发生降解,因此本方法及所有基于酸水解的分析技术均无法检测该组分。
在ACQUITY Premier系统上实施AOAC方法2018.06
配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统,其分散体积、梯度延迟体积和系统驻留体积与其他ACQUITY UPLC系统不同。实施AOAC方法2018.06时,需要调整色谱条件和步骤,以实现可靠、高效的分离。色谱条件和步骤的主要调整包括:改进了流动相A制备步骤、使用6倍溢出的PLNO进样模式、使用在线过滤器,并对衍生化配方进行了微调。
流动相A的制备
研究发现,保留时间(RT)小于10分钟的早期洗脱AA对初始流动相A的组成非常敏感。新鲜制备的流动相A批次不同,通常会导致这些化合物的RT发生明显漂移。为减少RT波动,我们改用A级容量瓶替代量筒来移取AccQ∙Tag Ultra洗脱液A浓缩液(请参阅“实验”部分的“UHPLC分离条件”)。此外,在制备流动相A时,将AccQ∙Tag Ultra洗脱溶液A浓缩液和水的体积比从150:850调整为100:600 (v/v)。体积比的调整略微增加了AA的RT,有效避免了因流动相A配制的微小差异导致先洗脱的AA (His)与衍生化峰(在在His之前洗脱)的分离度下降问题。
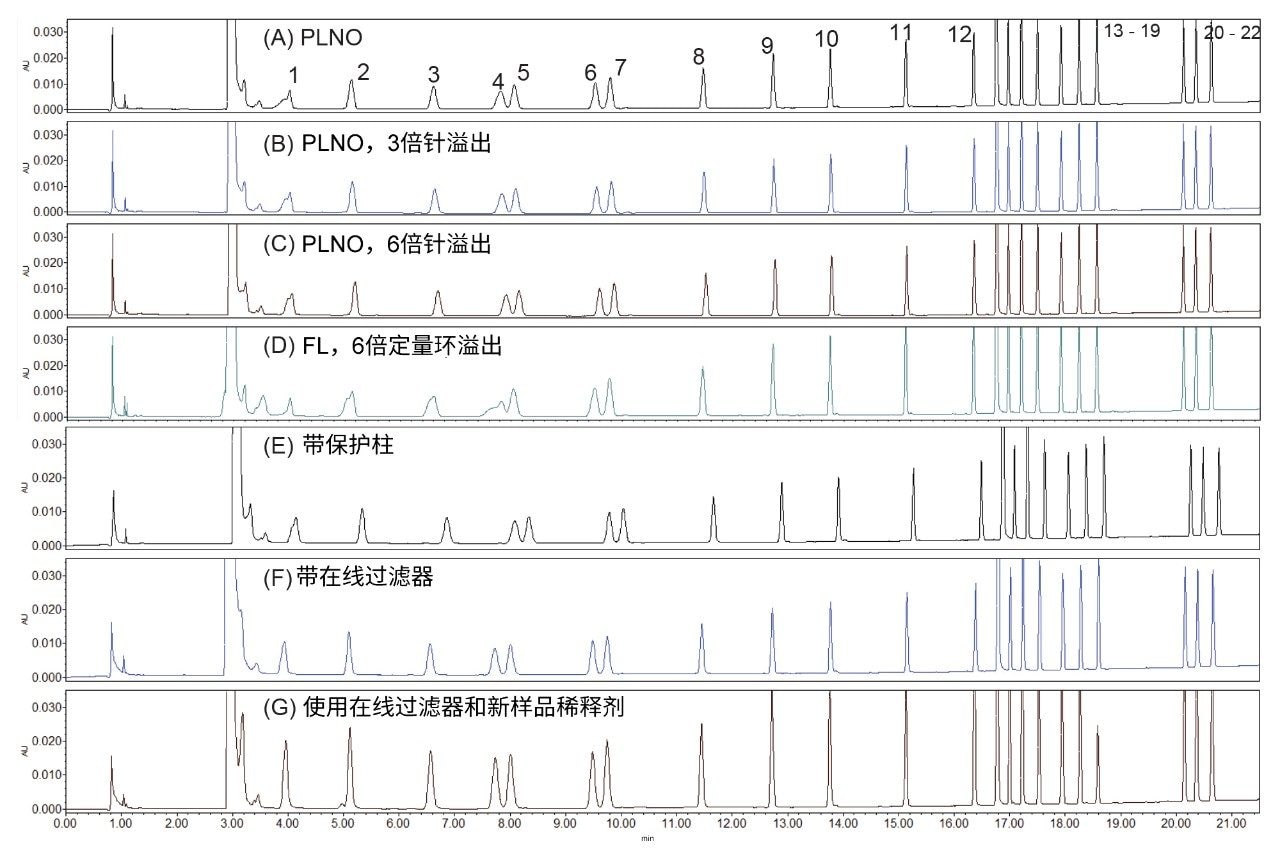
改善峰形
早期洗脱AA的峰形(图1)存在偏斜。这可能是由于样品稀释剂中的有机相含量明显高于初始流动相,从而引发了强溶剂效应。本研究尝试了多种优化峰形的方法。图1展示了不同条件对AA峰形的影响。这些条件包括不同的进样模式(针溢出体积比)、使用色谱柱在线过滤器或保护柱,以及调整降低样品稀释剂有机相含量。获得理想峰形的组合条件为:PLNO进样模式(针溢出体积比为6)、使用在线过滤器、降低样品稀释剂中的有机相含量(在衍生化步骤中将硼酸盐缓冲液从70 µL改为 100 µL)。
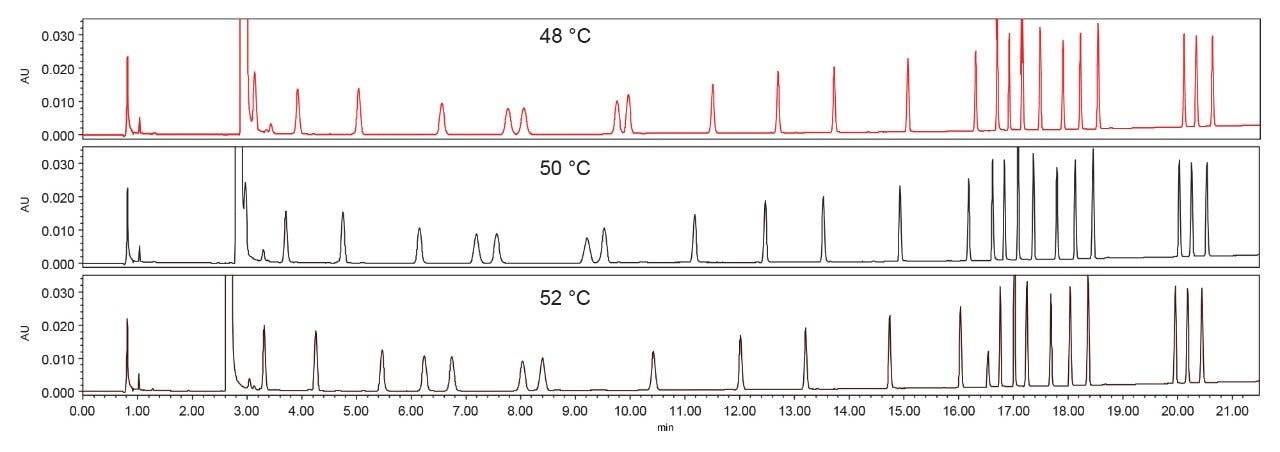
柱温
柱温也是优化AA分离效果的关键因素。图2展示了柱温对AA的RT和选择性的影响。即使柱温仅发生细微变化(± 2 °C),也会导致AA的保留时间和选择性发生显著变化。最终确定50°C的柱温条件能够为AA提供适宜的保留时间和良好的分离度。
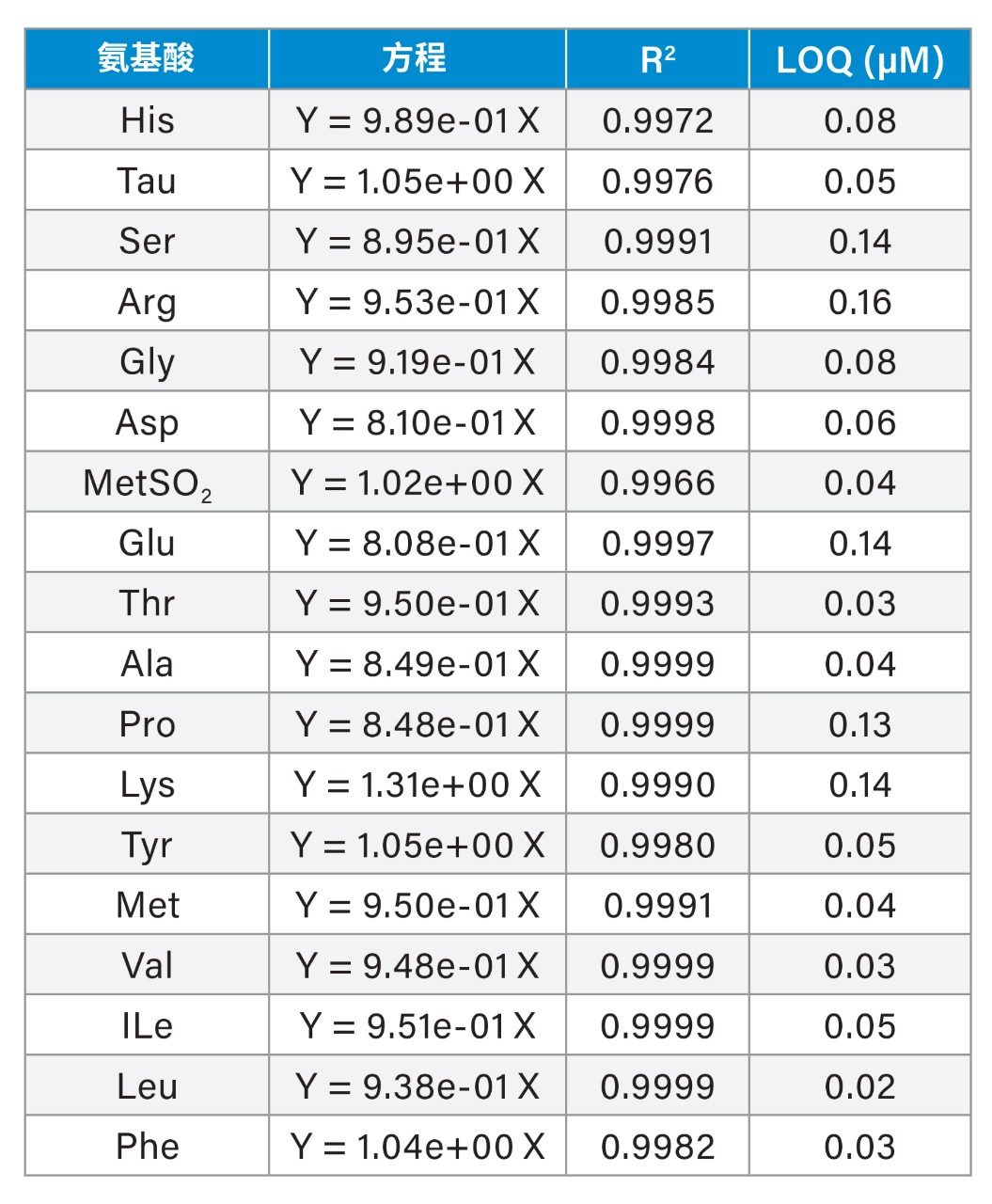
灵敏度和线性
采用0~19.2 µM浓度范围的AA工作标准溶液进行线性评估。通过最小二乘回归将数据点拟合至过零线(未加权)。表2展示了每种AA的代表性标准曲线方程、确定系数(R2)和定量限(LOQ)估值。所有AA的R2均大于0.997。LOQ采用行业指南中所述方法,通过响应标准偏差和标准曲线斜率进行估算9。AA的响应值以AA与内标(Nva)的峰面积比值乘以内标浓度表示。LOQ估值基于0.19 µM AA混标溶液的五次进样结果。LOQ估值范围为0.02~0.16 µM(见表2)。
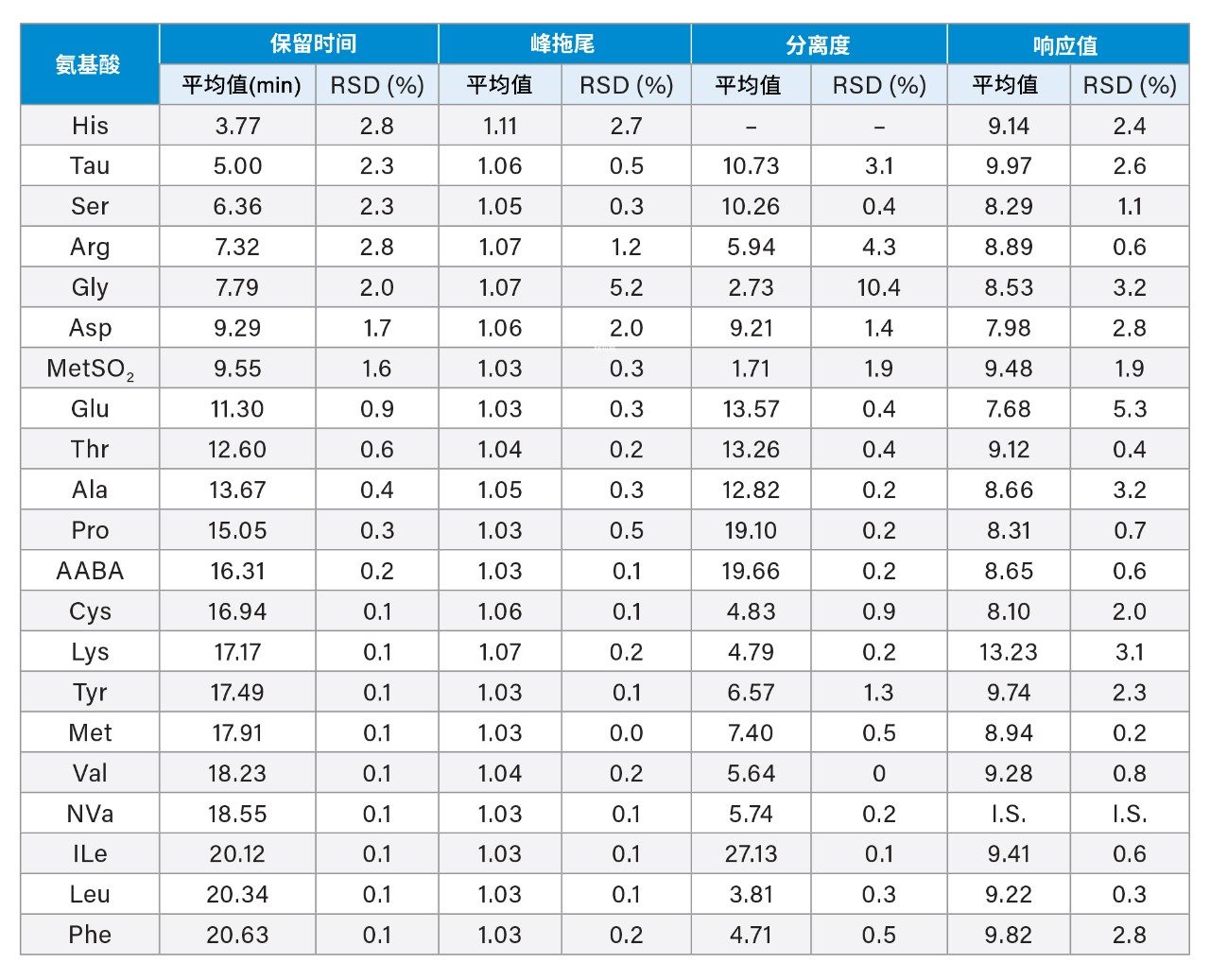
RP-UPLC的重复性和中间精度
研究通过重复进样(n=4,一天内测定)食品和饲料AA混标溶液(7.7 µM),评估了RT、峰拖尾、分离度和响应方面的重复性。表3所示为每种AA的RT、峰拖尾、分离度和响应的平均值及RSD值。早期洗脱化合物(RT < 10 min)的RSD值小于3%,其他化合物的RSD值小于1%。除His(最早洗脱的AA)峰拖尾为1.11外,其余所有AA的峰拖尾均小于1.07。除MetSO2的分离度为1.71(Asp与MetSO2之间)外,其余化合物的分离度均大于2.0。请注意,MetSO2并未列入AOAC 2018.06的AA列表中,且在乳制品样品中未发现(该化合物包含在沃特世食品和饲料AA混标中)。Glu的AA响应值RSD为5.3%,其余AA均低于3.2%。
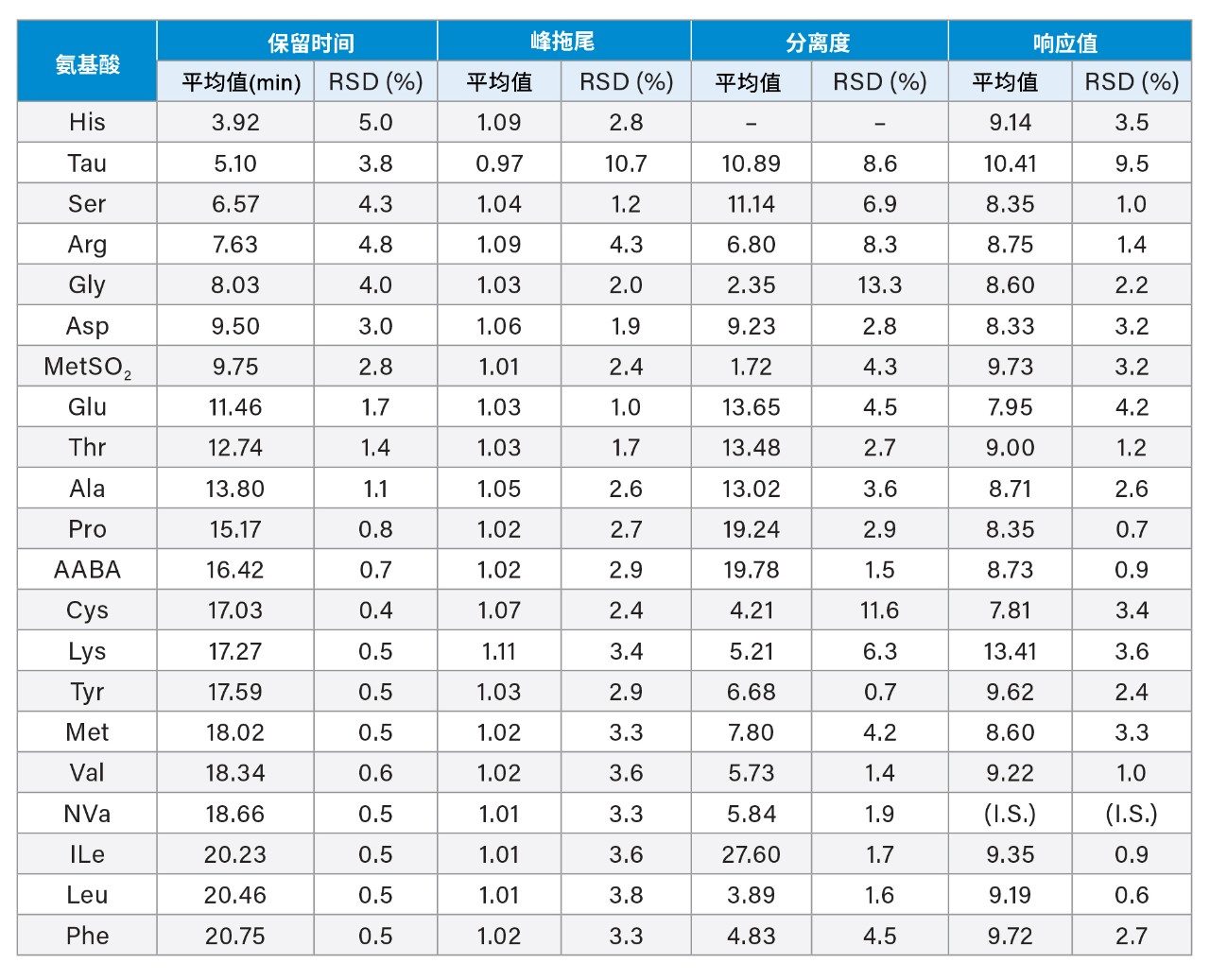
研究还评估了中间精度,使用单独制备的食品和饲料AA混标溶液(7.7 µM),在不同的3天使用3个不同批次的色谱柱进行分析。表4展示了汇总结果。早期洗脱AA (RT < 10 min)的RT RSD小于5.0%,其余AA的RT RSD小于1.7%。所有AA的峰拖尾均小于1.11。除MetSO2(MetSO2与Asp对)的分离度为1.72外,其余化合物的分离度均高于2.0。各AA响应值的RSD中,Tau为9.5%,其他AA均低于4.2%。
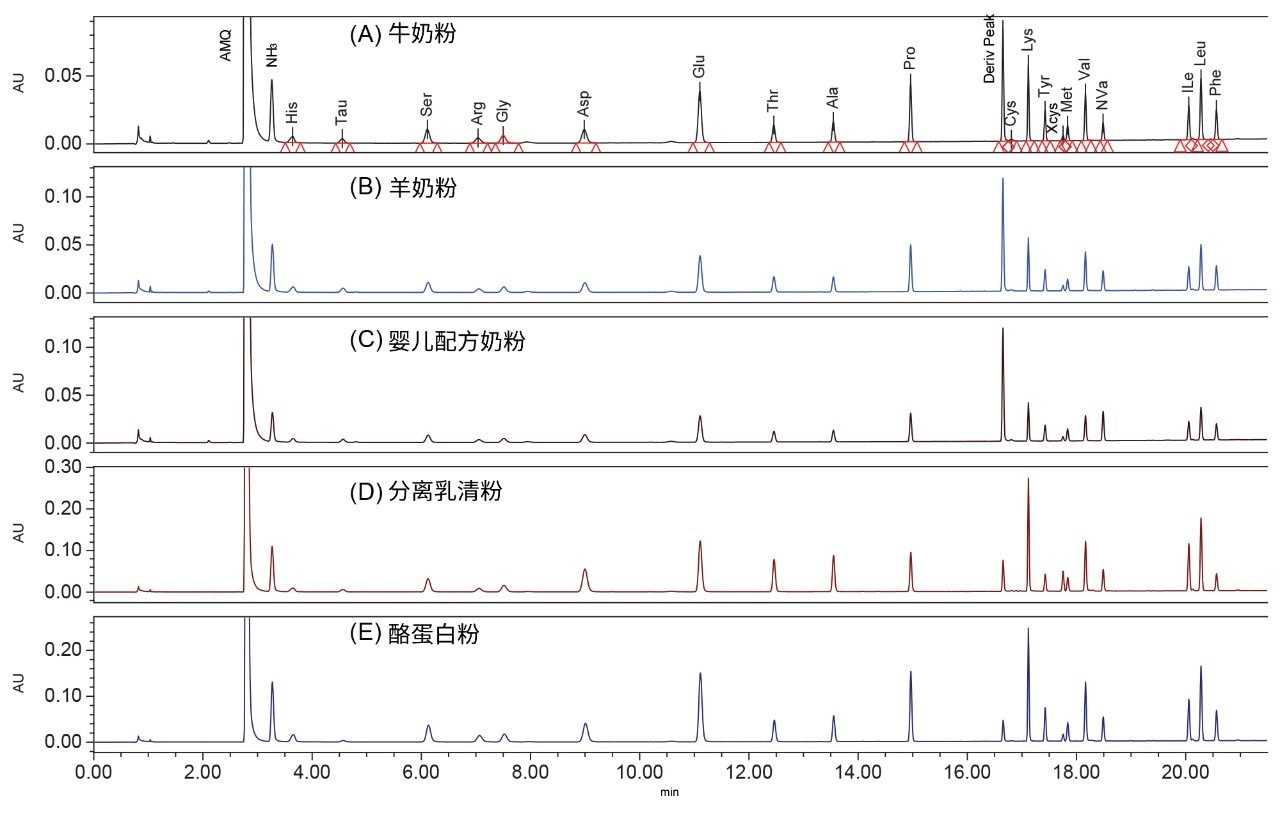
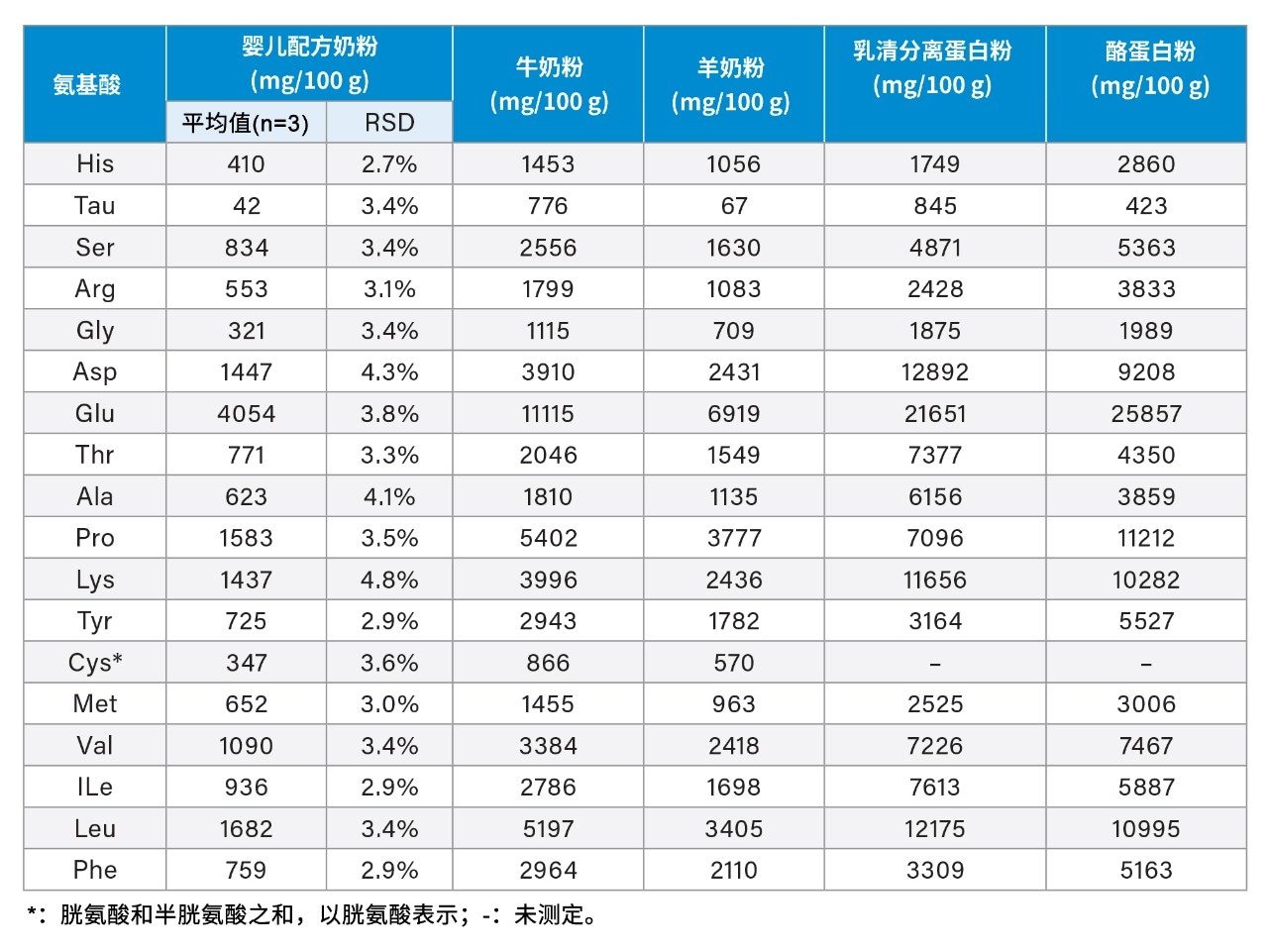
乳制品分离分析
乳制品中的所有AA均实现了基线分离(图3)。检测的乳制品样本包括牛奶粉、山羊奶粉、婴儿配方奶粉、乳清分离蛋白粉和酪蛋白粉。表5展示了这些乳制品中测得的AA含量。婴儿配方奶粉经三次平行测定,其AA结果的RSD均低于4.8%。
结论
本研究采用配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统,结合AccQ∙Tag Ultra C18色谱柱(1.7 µm, 2.1 mm × 150 mm),成功建立了乳制品中AA的检测方法。分析步骤在遵循AOAC方法2018.06的基础上进行优化改进,包括改进流动相A制备程序以获得更一致的AA RT、优化色谱条件(进样模式/参数及使用在线过滤器),以及改良衍生化配方以获得更出色的峰形。采用符合AOAC方法要求的Waters AccQ∙Tag Ultra消耗品(衍生化试剂盒、标准品和化学品试剂盒及色谱柱)成功完成了该分析。使用ACQUITY Premier系统在改良的AOAC方法条件下分离AA,展现出优异的线性、灵敏度和重复性(包括中间精度)。所有AA均获得了出色的峰形和分离度。对婴儿配方奶粉、牛奶粉、山羊奶粉、乳清蛋白粉和酪蛋白粉等常见乳制品样本中的AA分析表明,该系统具有优异的分析性能。配备二元溶剂管理器和固定定量环样品管理器的ACQUITY Premier系统与AccQ∙Tag Ultra化学品试剂盒相结合,为乳制品中AA的检测提供了可靠的解决方案。
参考资料
- Jaudzems, G.; Fuerer, C. Determination of total amino acids in infant formulas, adult nutritionals, dairy, and cereal matrixes by UHPLC–UV: Interlaboratory Validation Study, Final Action 2018.06. Journal of AOAC International 2022, 105 (6), 1625–1639.
- International Organization for Standardization.(2022).Milk and milk products – Determination of amino acids in infant and adult/paediatric nutritional formulas and dairy products (ISO Standard No.4214:2022).
- Cereals and Grains Association.(2022).Total Amino Acids by UHPLC-UV (AACC 07-50.01).
- UPLC Amino Acid Analysis Solution System Guide.Waters Corporation. 71500129702 Revision B, 2007.
- Amino Acid Analysis Application Notebook.Waters Corporation.720006130, 2018.
- 关于多实验室测试中使用的Waters ACQUITY系统清单,请查阅参考资料1中的附表1。
- AccQ∙Tag Ultra Derivatization Kit Care and Use Manual.Waters Corporation.715001331 Rev. A, 2024.
- Cohen, S. Analysis of Sulfur Containing Amino Acids III.Alkylation of Cysteine.Waters Lab Highlights.LAH0379, 1988.
- ICH.(2005) Q2(R1) Validation of Analytical Procedures: Text and Methodology Guidance for Industry.(Accessed on June 2nd, 2023).
720008632ZH,2024年12月