HILIC UV/MS法分析寡核苷酸治疗药物(包括siRNA双链体、脂质偶联ASO和CRISPR sgRNA)的流动相研究
摘要
亲水作用液相色谱(HILIC)已成为一种有望替代传统离子对反相(IP-RP)方法用于寡核苷酸(ON)分离的方法。本研究探讨了HILIC在分离ON非对映体、双链体和单链组分方面的应用,强调了该技术与质谱(MS)的兼容性,因此无需使用离子对试剂。我们采用实验设计(DoE)系统地优化了离子强度、温度和流动相组成等关键参数,旨在提高灵敏度。本文还提出了使用连续进样方案消除流穿效应和峰分裂。
优势
- HILIC是一种无需使用离子对试剂的色谱方法,可轻松搭配不同浓度的铵类流动相使用
- 虽然其电离效率低于使用HFIP的IP-RP LC-MS,但该LC-MS方法在流动相离子强度低至25 mM时依然可靠
- GTxResolve™ Premier BEH™ Amide 300 Å 1.7 µm色谱柱经过批次测试,因其在分离核酸和蛋白质方面的出色性能而被选择
- MaxPeak™ Premier色谱柱硬件可提高回收率,并且尽可能减少使用前的色谱柱钝化需求
简介
寡核苷酸分离的HILIC方法
近年来,亲水作用液相色谱(HILIC)在寡核苷酸(ON)分离方面的应用日益受到关注1,2,3。 与目前广泛使用的离子对反相(IP-RP)方法相比,HILIC模式提供了一种灵敏、稳健且有吸引力的替代方案。研究发现,HILIC在ON非对映体分离中展现出独特优势。高离子强度和低温流动相条件可显著提高对映体的选择性3。 此外,HILIC可根据流动相温度灵活切换至变性或非变性(天然)条件,从而有效分离双链体与其单链组分4。 HILIC的另一个重要优势是能够使用不含离子对的流动相,因此备受MS应用青睐5,6。 目前,HILIC已成功应用于LC-MS及LC-MS/MS平台中sgRNA的自下而上测序7。
尽管针对ON开发的HILIC方法已具备较高稳定性,但其保留机制尚未完全阐明。部分学者认为离子相互作用(需注意的是此情况下应为排斥效应)可能参与保留过程,而另一些研究则指出HILIC对ON的分离主要由氢键相互作用主导,具体机制可能涉及吸附或分配过程1,3。 一般认为,高离子强度的流动相能够稳定极性(即酰胺基)固定相表面的固定化水层,并大幅减少离子相互作用3。 然而,部分色谱模型提出了相反观点:当向水性流动相中添加盐时,水分子会优先溶剂化盐离子(破坏水的高度有序结构),从而减少可用于与亲水表面相互作用的水分子数量(预期会降低固定化水层的厚度)。另外值得注意的是,HILIC色谱柱在使用质子助溶剂(即甲醇)替代乙腈时,能保持良好的峰形和相似的选择性,这一现象进一步增加了理解保留机制的复杂性。最后需注意的是,从HILIC色谱柱洗脱ON所需的水相比例(20~60%)显著高于糖或糖蛋白的洗脱需求(15~35%)8。
实验
LC方法注意事项
目前针对HILIC在ON分离中的系统性方法开发鲜有报道。研究者常采用相同或相似条件(10~50 mM醋酸铵、甲酸铵或碳酸氢铵搭配助溶剂乙腈)。虽然已对流动相添加剂的离子强度、温度效应及pH值展开研究,但多为单独研究(单因素实验法,OFAT),而非结合实验设计(DoE)或多维保留模型进行研究1,3,9。 本研究采用系统性方法比较了三种铵盐的影响。首先进行筛选,通过三因素DoE研究梯度倾斜度(梯度时间,tG)、温度(T)和离子强度(C)的影响。其中,tG和T在两个水平下研究,C在三个水平下研究。然后,使用DryLab™ 4.4软件(Molnar Institute,德国柏林)同步优化梯度程序和温度,获得可普遍应用于不同寡核苷酸ladder样品的10分钟分离方案。
由于样品稀释剂(蛋白质或寡核苷酸等生物分子必须溶于水相)与流动相(主要为非质子溶剂乙腈)的固有不相容性,HILIC分离过程中常出现流穿效应(或部分峰分裂)10。 色谱分析工作者常采取折中方案:用乙腈-水混合液稀释样品以避免流穿效应,但这可能导致样品沉淀和/或变性。本文提出一种更巧妙的解决方案:在纯水相溶剂塞进样的前后分别注入弱溶剂(乙腈)塞(即顺序化或“括号式”进样法)。
寡核苷酸标准品/Ladder的LC分析
样品前处理:
将一瓶MassPREP™寡核苷酸标准品(P/N:186004135)内容物复溶于100 μL水中,制备寡核苷酸dT ladder。将一瓶ssDNA Ladder 20–100(P/N:186009448)内容物复溶于100 μL水中,制备ssDNA ladder。将脂质偶联ASO LC-MS标准品(P/N:186010747)复溶于20 μL丙酮中,再用80 μL水稀释,制备脂质偶联ASO寡核苷酸样品。将siRNA LC-MS标准品(P/N:186010598)复溶于100 μL水中,用于研究HILIC中的解链温度。
流动相制备:
HPLC级水和乙腈(MeCN)购自Fisher Scientific(爱尔兰都柏林)。醋酸铵、甲酸铵和碳酸氢铵购自Sigma-Aldrich(瑞士布克斯)。在LC研究中,为避免沉淀和溶解度问题,采用了预混合流动相。流动相A为水/乙腈(30/70),流动相B为水/乙腈(90/10)。研究发现,采用上述流动相组成时,通过运行0-100%B的线性梯度,所有寡核苷酸标准品均能以适当的保留时间洗脱。为研究流动相中添加剂的影响,本研究考察了三种铵盐在流动相A和B中的三种离子强度(C1 = 10 mM,C2 = 25 mM,C3 = 50 mM)。
在最终优化的LC-UV方法中,流动相A和B均添加了50 mM盐。
其他实验条件(LC-UV)如下表所示:
|
液相色谱系统: |
配备二元溶剂管理器的ACQUITY™ UPLC ™ H-Class Bio系统[相当于配备BSM FTN仪器的ACQUITY Premier系统,并配置高pH试剂盒] |
|
检测: |
ACQUITY TUV检测器(钛合金流通池,5 mm,1500 nL) |
|
波长: |
260 nm |
|
数据采集: |
Empower™ Pro 3软件Feature Release 3 |
|
色谱柱: |
GTxResolve Premier BEH Amide色谱柱, 300 Å, 1.7 µm, 2.1 x 50 mm(P/N:186011249) |
|
柱温: |
优化后的方法为50 °C(分别设定为30 °C和70 °C以模拟温度效应) |
|
样品温度: |
6 °C |
|
样品瓶: |
采用MaxPeak™ HPS技术的QuanRecovery™ 12 × 32 mm螺纹口样品瓶,300 μL(P/N:186009186) |
|
进样体积: |
0.5-1 µL |
|
流速: |
0.4 mL/min |
|
流动相: |
A:50 mM醋酸铵/甲酸铵/碳酸氢铵,溶于水/乙腈(30/70) B:50 mM醋酸铵/甲酸铵/碳酸氢铵,溶于水/乙腈(90/10) |
|
注射器吸取速度: |
30 µL/min |
|
进样针位置: |
1.0 mm |
|
TUV采样速率: |
20 Hz |
|
滤波器时间常数: |
无 |
脂质偶联ASO和CRISPR sgRNA的LC-MS分析
将沃特世脂质偶联ASO LC-MS标准品(P/N:186010747)复溶于20 μL丙酮中,再用80 μL无核酸酶的水稀释。HPRT sgRNA购自IDT,将其溶于无核酸酶的水中,浓度为50 pmol/μL。
|
液相色谱系统: |
BioAccord™ LC-MS系统 |
|
检测: |
ACQUITY TUV检测器(分析型流通池) |
|
波长: |
260 nm |
|
数据采集: |
Empower Pro 3 Feature Release 3 |
|
色谱柱: |
GTxResolve Premier BEH Amide色谱柱, 300 Å, 1.7 µm, 2.1 x 50 mm(P/N:186011249) |
|
柱温: |
75 °C |
|
样品温度: |
6 °C |
|
样品瓶: |
采用MaxPeak HPS技术的QuanRecovery 12 × 32 mm螺纹口样品瓶,300 μL(P/N:186009186) |
|
进样体积: |
1 µL |
|
流速: |
0.4 mL/min |
|
流动相: |
A:100 mM醋酸铵,溶于18.2 MΩ水/1%乙腈,使用IonHance醋酸铵浓缩液(P/N:186009705)制备,盛装在1 L经认证的LDPE储液瓶(P/N:186009110)中 |
|
(sgRNA): |
B: LC-MS级乙腈,盛装在1 L经认证的玻璃储液瓶中(P/N:186007089) |
|
流动相: |
A:25 mM醋酸铵,溶于18.2 MΩ水/0.25%乙腈,使用IonHance™醋酸铵浓缩液(P/N:186009705)制备,盛装在1 L经认证的LDPE储液瓶(P/N:186009110)中 |
|
(脂质ASO): |
B: LC-MS级乙腈,盛装在1 L经认证的玻璃储液瓶中(P/N:186007089) |
|
注射器吸取速度: |
30 µL/min |
|
进样针位置: |
2.0 mm |
|
TUV采样速率: |
20 Hz |
|
滤波器时间常数: |
无 |
|
MS模式: |
全扫描 |
|
质量范围: |
高(400~5000 m/z) |
|
扫描速率: |
5 Hz |
|
锥孔电压: |
40 V |
|
脱溶剂气温度: |
550 °C |
LC-MS实验的梯度程序
结果与讨论
三种铵盐流动相添加剂比较
当C ≤ 40 mM时,离子强度的影响很大。当C ≤ 40 mM时,保留性随离子强度降低而降低。如果添加剂浓度过低(即C = 10–20 mM),即使首峰表观保留因子(即kapp ~ 5)看似较高,仍可能出现流穿效应和/或峰分裂。在高温下,峰分裂和流穿效应会更加严重。
总体而言,温度是调节不同ON的选择性甚至洗脱顺序的关键变量,因此建议针对给定样品优化温度条件。
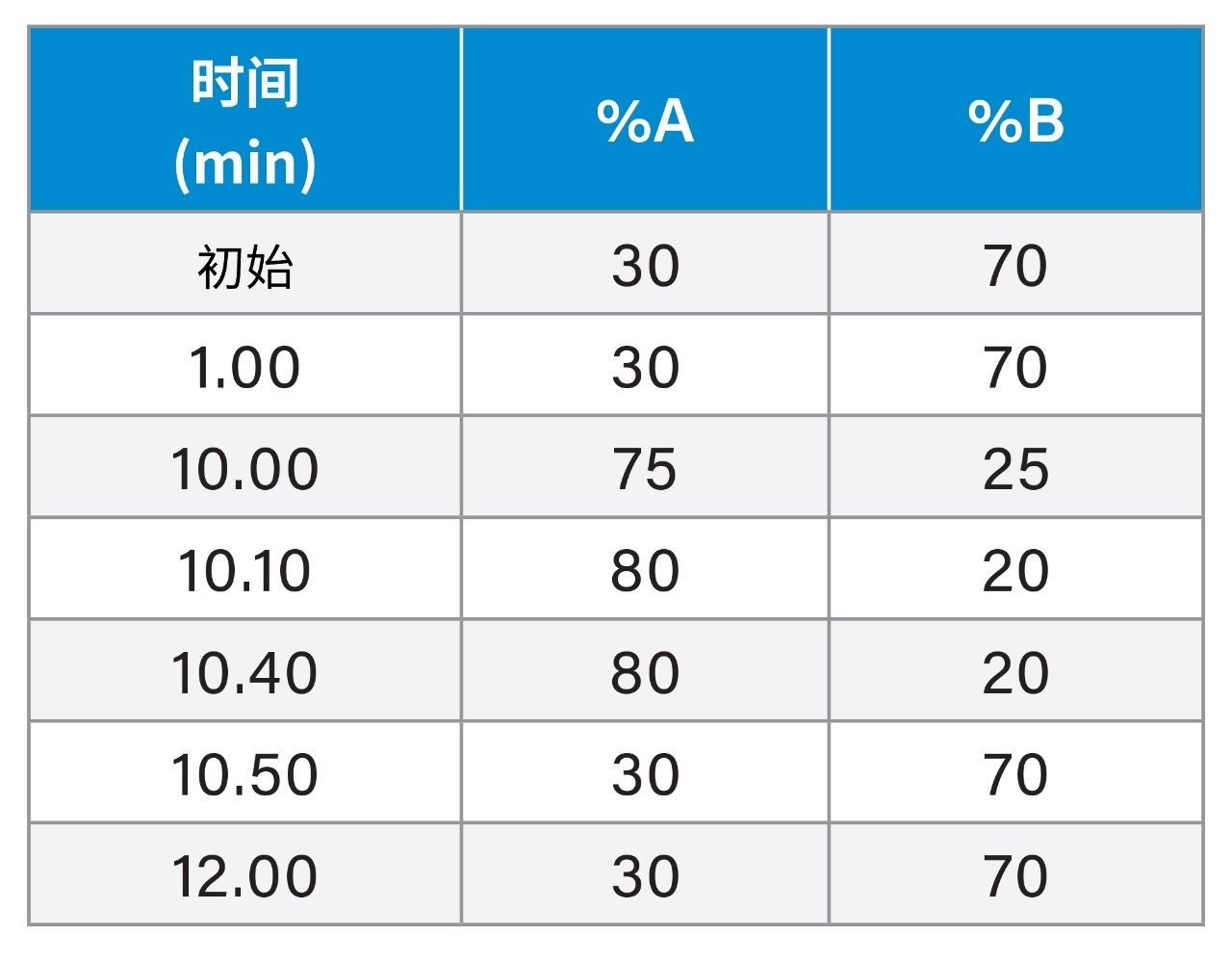
HILIC的梯度程序中也可以遵循与IP-RP相同的注意事项11。 由于ON具有同源性特征(即ladders),在运行线性流动相梯度时,其保留时间呈对数增长而非线性增长,因此采用对数或凹形梯度尽可能提高选择性。图1展示了三种盐(添加剂)在优化条件下(保持相同梯度倾斜度)的色谱图。
从色谱分离的角度来看,醋酸铵和甲酸铵具有等效性。这两种盐可采用相同的梯度程序,获得几乎一致的保留性和选择性。而碳酸氢铵的保留性低于醋酸盐或甲酸盐,因此必须调整梯度程序(偏移-5% B)才能获得相似的保留性。需注意的是,碳酸氢盐的选择性存在细微差异。相较于其他两种盐,使用碳酸氢盐时,部分先洗脱的峰更容易出现峰分裂和峰流穿(尤其在高温条件下),这是一个缺点。不过,通过采用括号式进样可消除这种影响(见下一节)。
采用括号式进样避免流穿效应
在HILIC分析中,由于进样溶剂的洗脱强度高于流动相(即流动相组成与进样溶剂间存在溶剂不匹配),样品可能在色谱柱入口处无法充分聚焦,从而导致色谱分离问题10。 通过精细调节进样稀释剂(即提高样品稀释剂中的乙腈比例)可大幅减少此类不匹配问题。然而,受样品性质限制(可能导致变性、诱导翻译后修饰甚至不溶/沉淀),此类调节并非总能实现。溶剂不匹配通常会对色谱分离产生不利影响,轻则导致峰轻微展宽,重则引发峰形严重畸变、分裂甚至分析物流穿。流穿现象是指部分样品未与固定相充分相互作用即通过色谱柱,并在接近色谱柱死时间处洗脱,而另一部分峰则按正常保留时间洗脱。
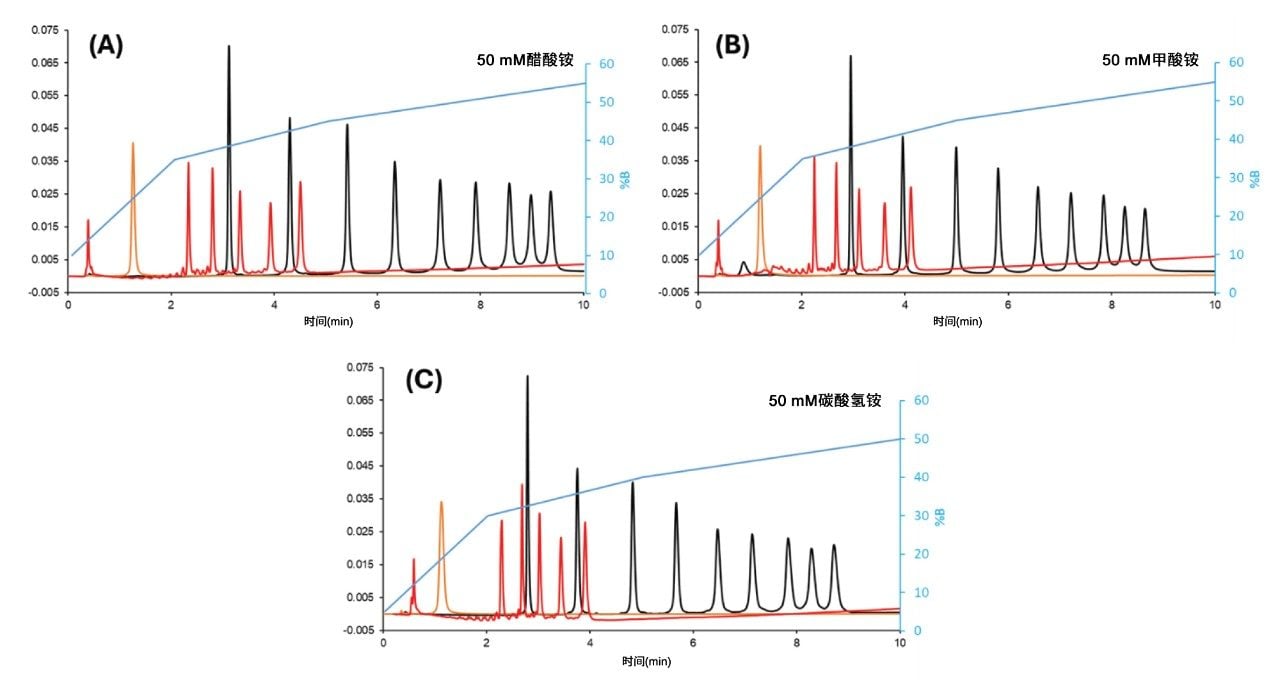
在本研究的示例中,寡核苷酸dT ladder对峰分裂最敏感。进样体积为0.5 μL时,第一种化合物(15 nt)洗脱出分裂峰。进样体积超过1.5 µL后,五种化合物(15、20、25、30和35 nt)均出现明显分裂,部分溶质在色谱柱死体积处洗脱。
括号式进样序列有助于消除流穿效应。该方法通过将样品夹在两段弱溶剂(本例中为乙腈)进样之间,使样品注入柱头。采用括号式进样后,针对dT ladder样品的进样体积可提升至2 µL而不出现分裂。图2展示了向流路中进样1 µL样品时常规进样与括号式进样的对比效果。
温度作为调节分离的关键参数
除调节不同物质的选择性和洗脱顺序外,温度在HILIC中设定非变性或变性分离条件时也至关重要。
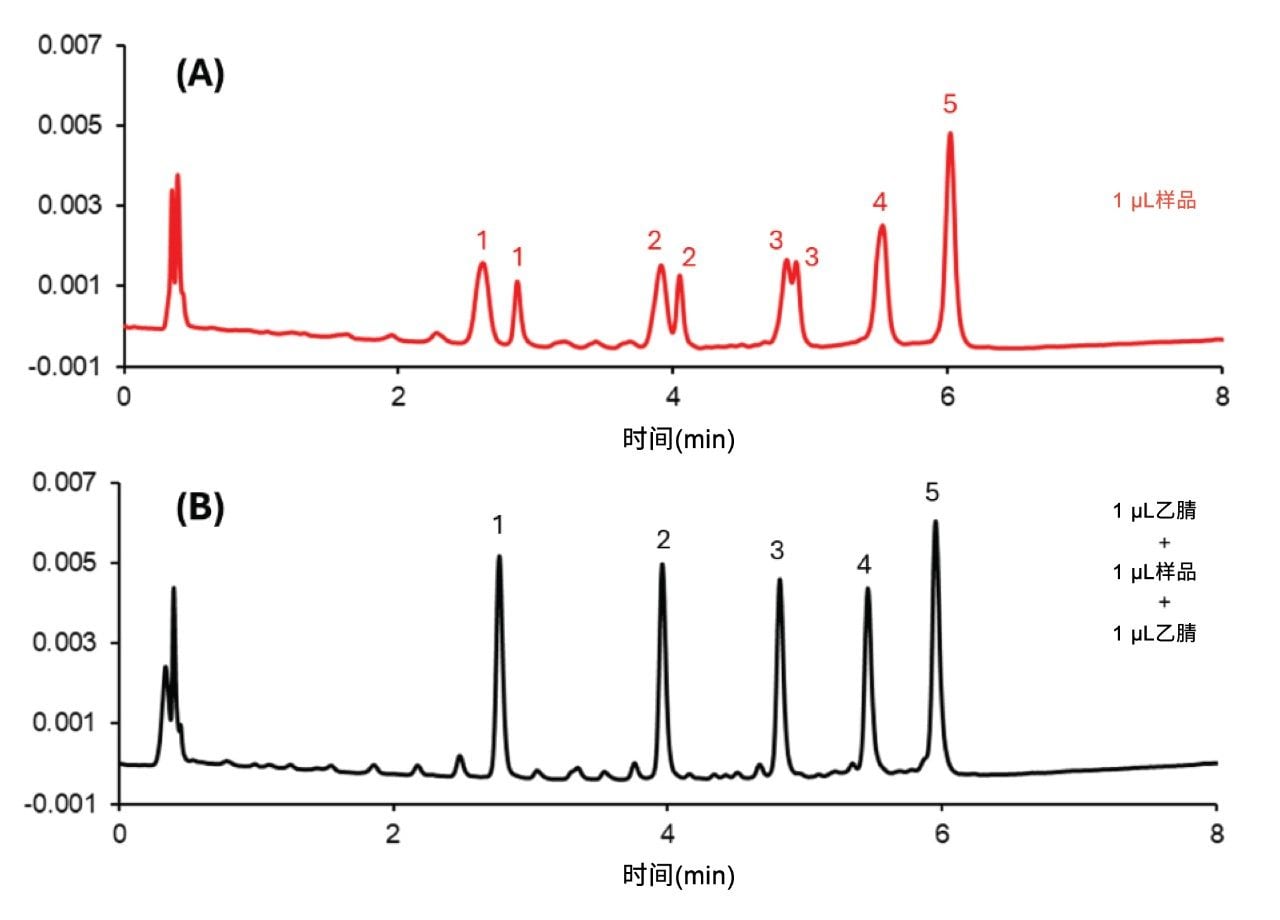
在低于双链体解链温度(Tm)的非变性条件下,ON双链体可以保持稳定。解链温度是衡量双链体稳定性的关键指标;但色谱条件如何影响双链体稳定性尚未完全明确4。 压力、温度和助溶剂可显著改变表观解链温度。
本研究使用siRNA标准品(退火的25 mer和27 mer RNA链的混合物)研究温度对双链体稳定性的影响。低于55 °C时,只观察到单峰。60 °C时,色谱图出现弥散宽峰,且保留时间较尖锐单峰显著缩短(数据未展示)。当温度升至65 °C以上时,色谱图上出现两个尖峰。这些结果表明,这种特定的siRNA在60 °C下开始解链成单链。图3显示了温度对siRNA双链体稳定性的影响。
CRISPR sgRNA和脂质偶联反义寡核苷酸的HILIC-MS分析
基于对寡核苷酸HILIC方法可调性的研究,进一步将该分离技术与质谱检测直接联用。本研究采用BioAccord LC-MS系统分析CRISPR sgRNA分子以及脂质偶联ASO(即脂质偶联ASO LC-MS标准品)。实验采用了两种不同的流动相组成。分析sgRNA时,使用经100 mM醋酸铵改性的水性洗脱液。分析脂质偶联ASO时,则使用更低浓度(25 mM)的醋酸铵。两种条件下均能产生有效水平的离子信号,且可通过单同位素峰或原始质谱图的MaxEnt去卷积处理获得的精确质量测定结果直接解析。
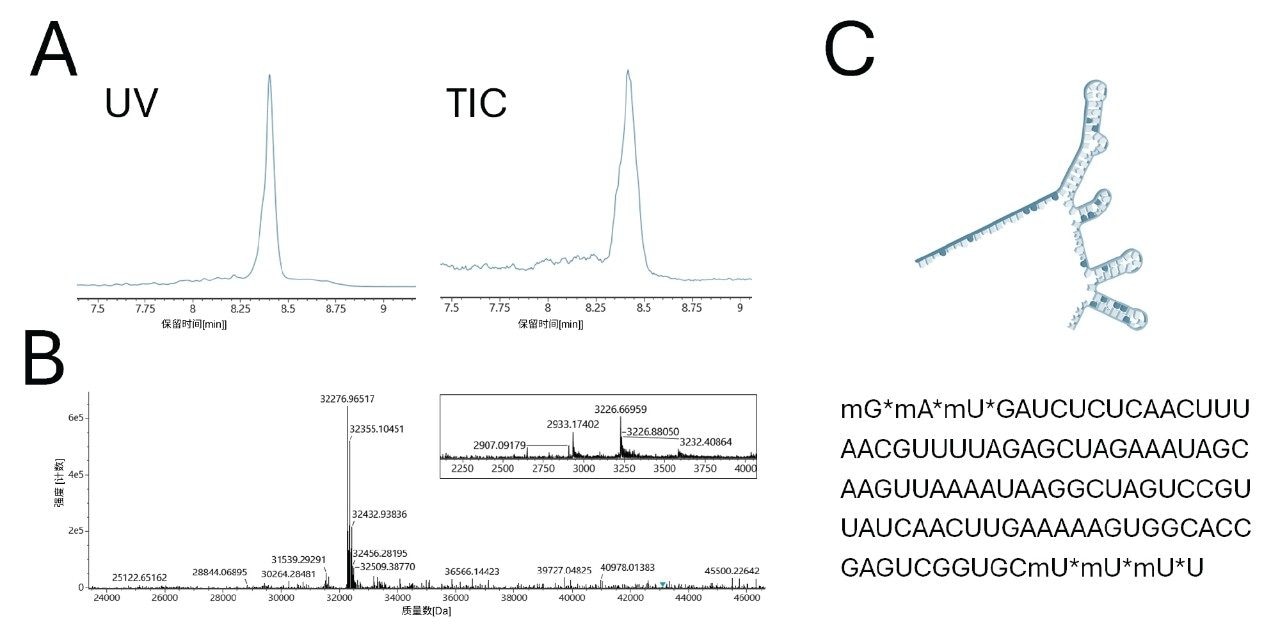
图4展示了利用人HPRT序列互补性合成的CRISPR sgRNA的LC-MS数据。在保留时间8.4分钟处观察到一个峰。该峰对应的MS信息如图B所示。插图显示了累加的原始质谱图以及一组[M-11H+]11-和[M-10H+]10-电荷态离子。对该质谱图进行MaxEnt去卷积处理后,得到sgRNA的完整分子量为32,276.97 Da,与包含5’和3’修饰末端的sgRNA序列理论值高度吻合。
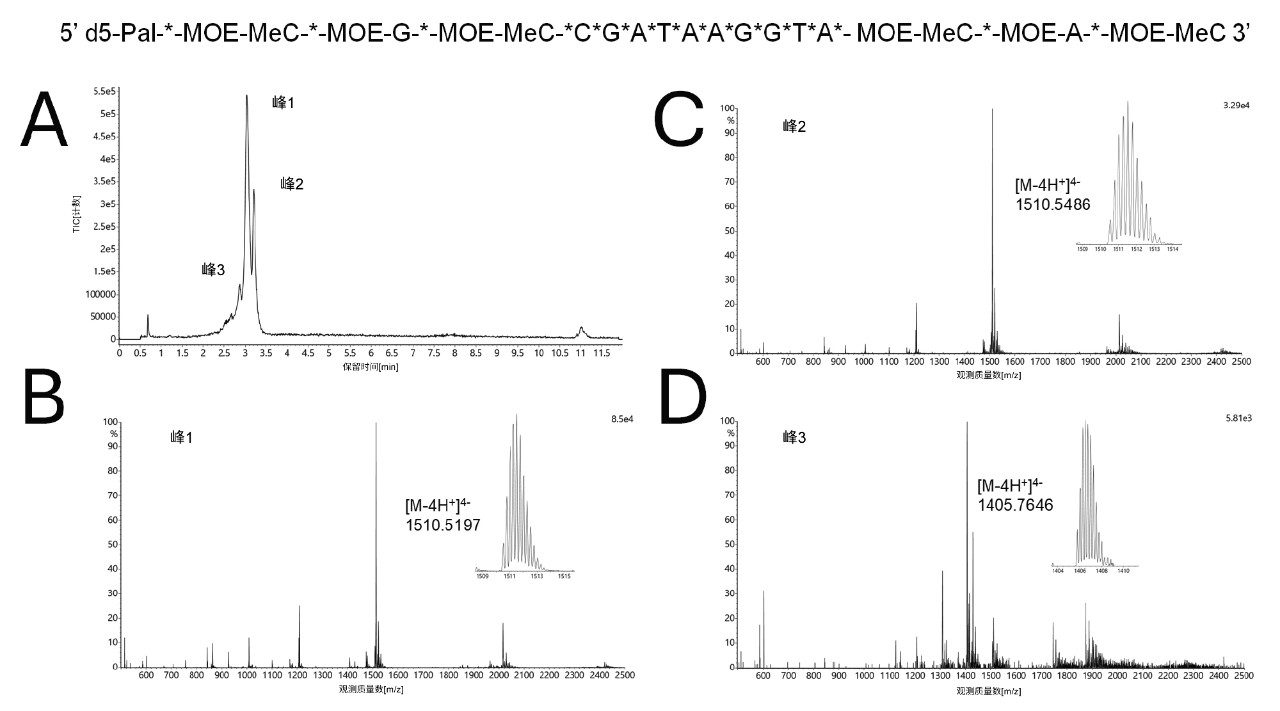
脂质偶联ASO的HILIC-MS分析结果见图5。与sgRNA相反,脂质ASO在HILIC色谱柱上的保留较弱。这归因于其明显较小的寡核苷酸尺寸(仅与C16脂质偶联)。该脂质ASO含有全硫代磷酸骨架及其他多种修饰。研究发现,采用高温分离可减少样品中对映体的峰分裂。在最终方法中(使用25 mM醋酸铵流动相),成功分离了两种同分异构体(峰1和峰2)。此外,还分离出一个质量数相差419 Da的样品杂质。
结论
本研究的结果证明HILIC方法用于寡核苷酸分离和分析的多项优势,并且在改进寡核苷酸治疗药物行业的分析方法方面表现出巨大潜力。
- 分离条件的灵活性:HILIC可以在变性和非变性条件下操作,能够分离双链体和单链寡核苷酸,这对于分析复杂的RNA结构和修饰至关重要。
- 与质谱的兼容性:HILIC能够使用不含离子对试剂的流动相,增强了其与MS的兼容性,提高了寡核苷酸测序和分析的灵敏度与准确度,例如在sgRNA的自下而上测序中。
- 高效方法开发和解决流穿效应:使用实验设计(DoE)可系统性优化温度、离子强度和梯度程序等关键参数,实现稳定、可重现的分离。括号式进样方法有效减少了样品流穿和峰分裂问题,能够在不影响样品完整性的前提下实现更高的进样体积和色谱性能。
HILIC是一种功能强大且通用的工具,可用于寡核苷酸的分离和分析,解决寡核苷酸治疗药物行业面临的重大挑战。随着业界对更精确、更灵敏分析方法的需求不断增加,尤其是在反义寡核苷酸(ASO)和RNA疗法等复杂治疗模式兴起的情况下,HILIC提供了一个稳定平台,确保高分离度、MS兼容性及方法开发的灵活性。其在质量控制、测序和杂质分析中的应用将在加速开发新一代寡核苷酸治疗药物中发挥关键作用。
Waters、MassPREP、ACQUITY、UPLC、Empower、GTxResolve、BEH、QuanRecovery、MaxPeak、BioAccord和IonHance是沃特世科技公司的商标。DryLab是Molnar Institute的商标。其他所有商标均归各自的拥有者所有。
参考资料
- M. Gilar, B.M. Koshel , R.E. Birdsall, J. Chromatogr.A 1712 (2023) 464475.https://doi.org/10.1016/j.chroma.2023.464475
- A. Goyon, M.S. Blevins, J.G. Napolitano, D. Nguyen, M. Goel, B. Scott, J. Wang, S.G. Koenig, T. Chen, K. Zhang, J. Chromatogr.A 1708 (2023) 464327.https://doi.org/10.1016/j.chroma.2023.464327
- H. Lardeux, K. Stavenhagen, C. Paris, R. Dueholm, C. Kurek, L. De Maria, F. Gnerlich, T. Leek, W. Czechtizky, D. Guillarme, M. Jora, Anal.Chem.96 (2024) 9994–10002.https://doi.org/10.1021/acs.analchem.4c01384
- M. Gilar, S. Redstone, A. Gomes, J. Chromatogr.A 1733 (2024) 465285.https://doi.org/10.1016/j.chroma.2024.465285
- P.A. Lobue, M. Jora, B. Addepalli, P.A. Limbach, J. Chromatogr.A 1595 (2019) 39–48.https://doi.org/10.1016/j.chroma.2019.02.016
- P. Anand, M. Koleto, D.R. Kandula, L. Xiong, R. MacNeill, Bioanalysis 14 (2022) 47–62.https://doi.org/10.4155/bio-2021-0216
- A. Goyon, B. Scott, K. Kurita, C.M. Crittenden, D. Shaw, A. Lin, P. Yehl, K. Zhang, Anal.Chem.93 (2021) 14792−14801.https://doi.org/10.1021/acs.analchem.1c03533
- B. Bobaly, V. D’Atri, A. Goyon, O. Colas, A. Beck, S. Fekete, D. Guillarme, J. Chromatogr.B 1060 (2017) 325–335.http://dx.doi.org/10.1016/j.jchromb.2017.06.036
- H. Lardeux, V. D’Atri, D. Guillarme, TrACs 176 (2024) 117758.https://doi.org/10.1016/j.trac.2024.117758
- R. Perez-Robles, S. Fekete, Róbert Kormány, N. Navas, D. Guillarme, J. Chromatogr.A 1713 (2024) 464498.https://doi.org/10.1016/j.chroma.2023.464498
- S. Fekete, M. Imiolek, M. Lauber, Platform Ion Pairing RPLC Method for Oligonucleotides Using High Throughput 20 mm Length Columns, Waters Application Note, 720008460, August 2024.
720008602ZH,2024年11月