使用ACQUITY™ QDa™ II质谱检测器简化阿托伐他汀中的杂质分析以实现的更高的检测和定量性能
摘要
- 展示QDa II质谱检测器在补充阿托伐他汀API杂质常规分析UV色谱工作流程方面的实际应用
- 评估分析工作流程的适用性,以确定符合ICH-Q3指南中≤0.15%的杂质鉴定阈值
- 强调源内裂解对于更深入地了解未知化合物结构特征的优势
- 重点介绍Empower™色谱数据系统(CDS)的用户友好特性和优势,适用于在受监管的制药环境中进行合规杂质检测
简介
阿托伐他汀是全球最广泛使用的处方药之一,用于控制高胆固醇血症等疾病和降低心血管事件风险。阿托伐他汀在降低LDL胆固醇水平方面的疗效使其成为了供给全球数百万患者的重要药物。然而,药物制剂中存在的杂质可能对患者安全构成严重威胁。这些杂质可能在活性物质的合成过程中产生,或者在某些环境条件下作为降解产物出现,并可能具有毒理学或药理学特性,会影响最终药品的安全性和有效性。
阿托伐他汀通常以活性羟酸的钙盐形式给药,剂量为10~80 mg/天1。 根据ICH-Q3指南,最大每日摄入剂量低于2.0 g的药物,其杂质的合格阈值应为0.15%2。
欧洲药典各论中关于活性物质阿托伐他汀规定了以下杂质限值:相关杂质A和B为0.3%;杂质C和D为0.15%;其他未指定的杂质为0.1%3。
传统的杂质检测方法,即色谱技术与UV检测联用,是制药分析中质量控制(QC)的基石。在该工作流程中辅以质谱检测,能够可靠且快速地鉴定、表征和定量分析杂质。此外,质谱检测通过提供额外的分子量信息,有助于在极低的检测限下识别杂质,从而扩展了分析的范围。
本应用纪要介绍了质谱检测在快速、准确地鉴定和定量分析阿托伐他汀API相关杂质方面的适用性。此外,我们还采用伪MS/MS方法通过源内裂解来表征杂质,从而提供一套全面的杂质分析解决方案。
实验
阿托伐他汀钙盐和三种相关杂质(杂质A、C和I)的市售样品购自Sigma Aldrich,并储存于4 °C下以备分析。将样品溶于甲醇(MeOH)中,用40:60的水:MeCN混合溶剂进行连续稀释以备分析。
LC-MS实验条件
|
LC系统: |
配备FTN-R样品管理器的ACQUITY UPLC™ H-Class系统 |
|
检测: |
ACQUITY QDa II质谱检测器 |
|
UV系统: |
光电二极管阵列(PDA)检测器 |
|
色谱柱: |
ACQUITY UPLC CSH™苯己基柱, 1.7 µm, 2.1 mm × 100 mm(P/N:186005407) |
|
柱温: |
30 °C |
|
样品温度: |
10 °C |
|
进样体积: |
2 µL |
|
流速: |
0.4 mL/min |
|
运行时间: |
27.5 min |
|
流动相A: |
10 mM醋酸铵水溶液 |
|
流动相B: |
含0.1%甲酸的乙腈溶液 |
|
样品瓶: |
12 × 32 mm透明玻璃螺纹颈口样品瓶(186000273) |
|
电离模式: |
正离子电喷雾(ES+) |
|
毛细管电压: |
1.1 kV |
|
脱溶剂气温度: |
600 °C |
|
离子源温度: |
120 °C |
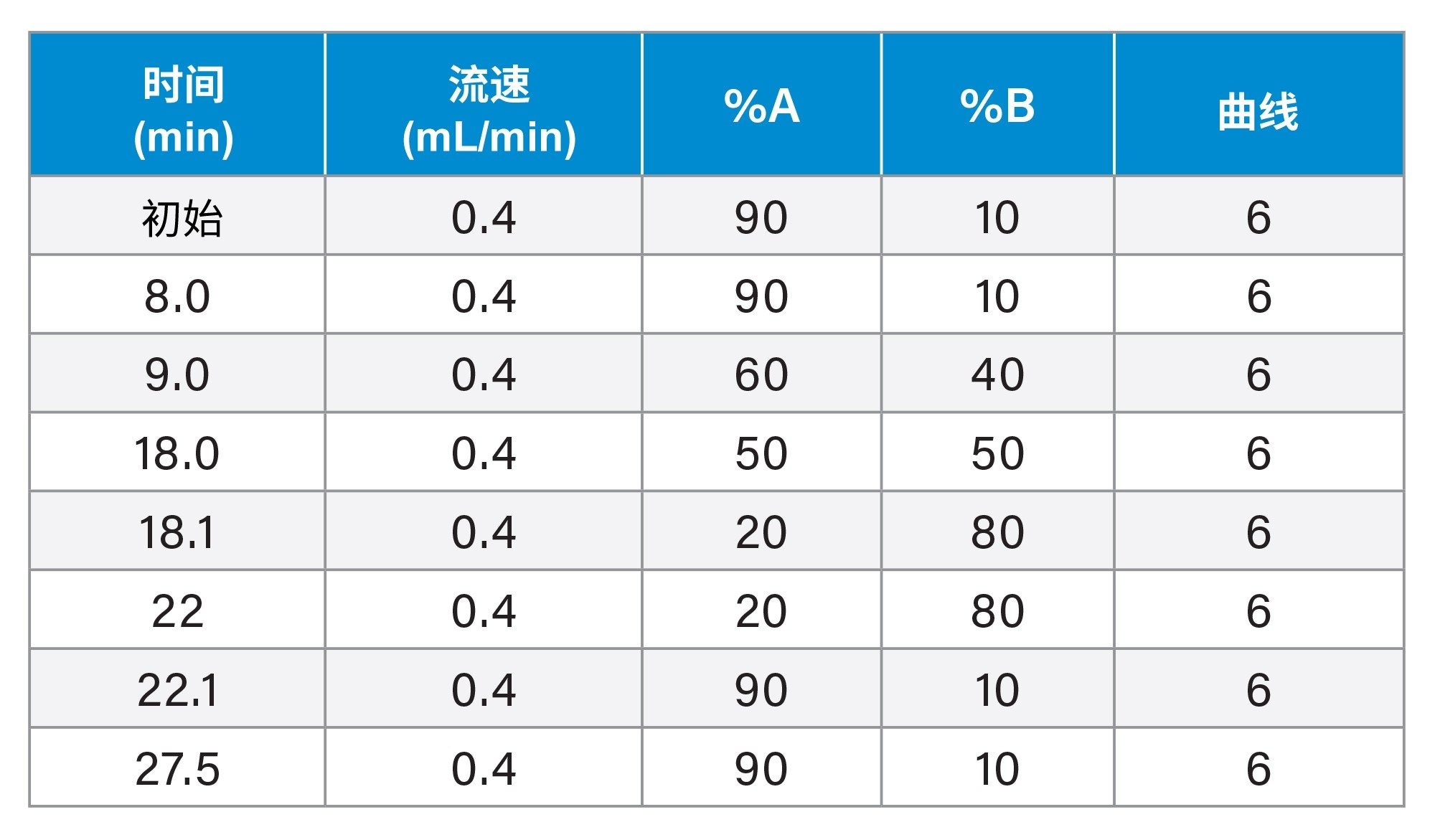
液相色谱梯度
软件
|
数据采集、处理和报告: |
Empower CDS 3.0 |
结果与讨论
方法开发
与欧洲药典各论中规定的方法不同,本应用纪要中介绍的分析方法在开发过程中并未使用四氢呋喃(THF)作为流动相,避免了毒性和不稳定的问题4。 而是在水相流动相中添加乙酸铵缓冲液,并在有机流动相中添加甲酸,来降低样品运行过程的pH并改善峰形。研究中还测试了几种LC色谱柱和流动相组成(包括将流动相pH优化至4.0~4.2),但与最终方法相比,色谱峰的分离度并没有提高5。 最终,该方法用时相较于欧洲药典专论方法大约缩短了60 min,且每次进样使用的溶剂体积不到10%。
使用1 µg/mL阿托伐他汀与选定杂质(A、C和I)的混合物对方法进行了优化。锥孔电压、毛细管电压和脱溶剂气温度均经过优化。尽管大多数文献方法都选择使用C18色谱柱,但CSH苯己基柱被证明是改善阿托伐他汀化合物与相关杂质C分离效果的理想选择。减少API峰拖尾的理想柱温为30 °C。
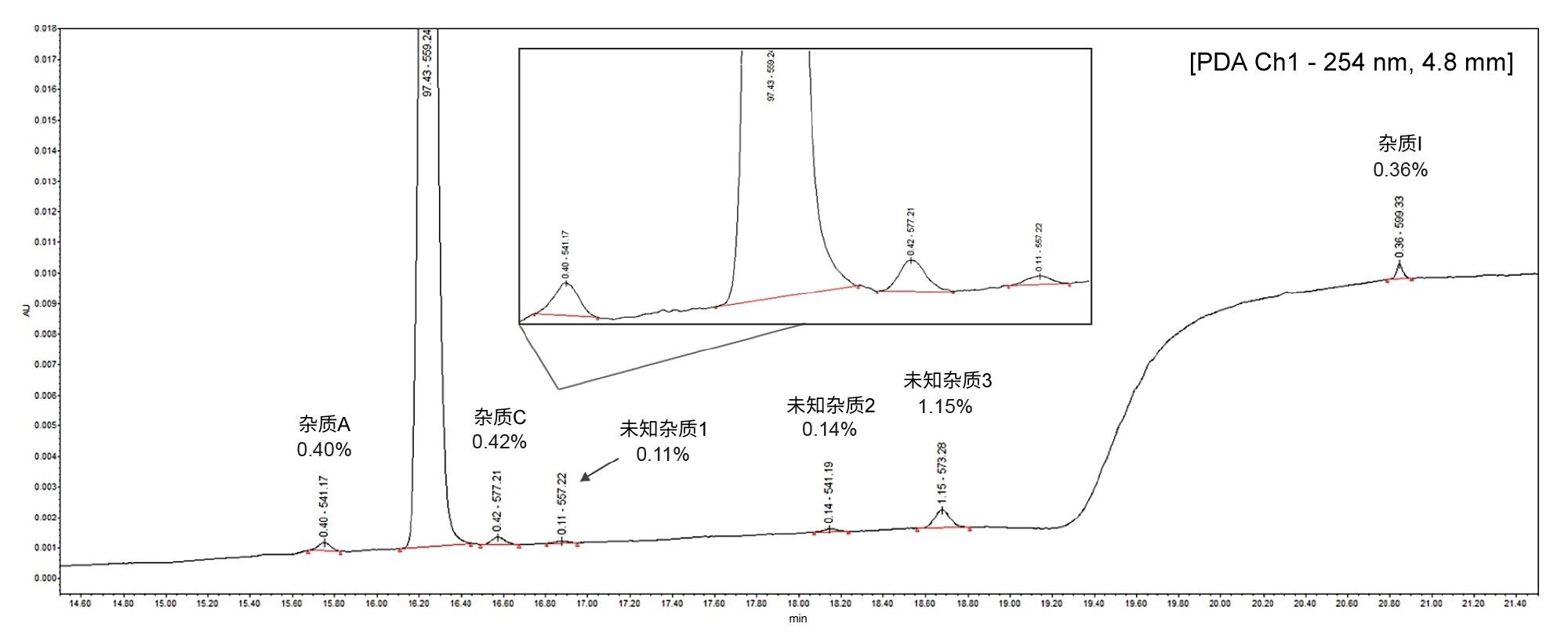
首先,向25 µg/mL的阿托伐他汀样品中添加选定的杂质以评估方法性能,杂质的浓度相对于API的浓度范围为0.36%~0.42%。在PDA迹线中还对三种未知杂质进行了定量,在图1中标记为未知杂质1、2和3,含量分别为API的0.11%、0.14%和1.15%。
线性
为了评估线性,研究中对API和杂质混标(含杂质A、C和I)进行了连续稀释。所有标准曲线均使用所述方法采集,总运行时间为27.5 min。
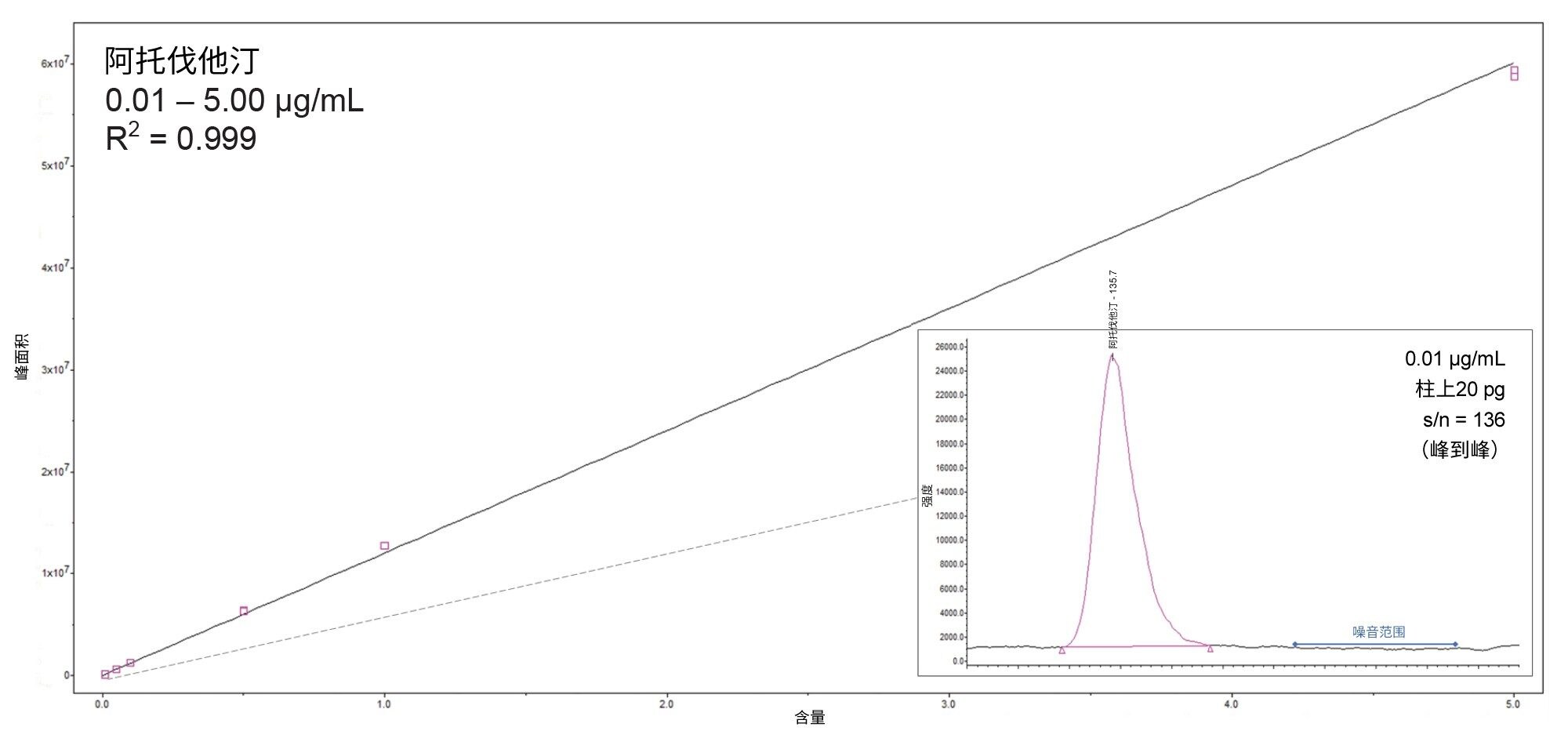
阿托伐他汀的间插标准曲线如图2所示。
- 该化合物在0.01~5.0 µg/mL的浓度范围内呈线性(1/X加权),R2值为0.999
- 在所有情况下,间插标准曲线的实测值百分比(%残差)均< 15.4%
- 最低校准点的峰(0.01 µg/mL,相当于20 pg柱上进样量),信噪比为136(峰到峰)
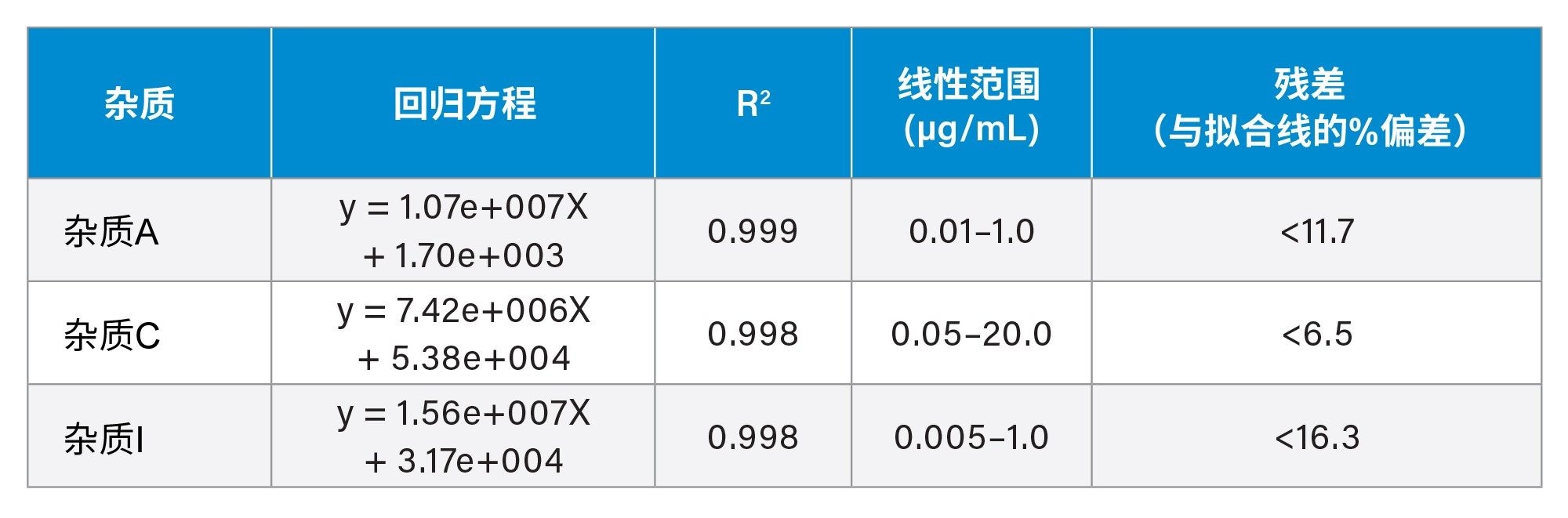
本研究还评估了选定的阿托伐他汀相关杂质的线性和灵敏度(表1)。所有情况下的R2值均超过0.998,且所有情况下的残差百分比均< 16.3%。所有标准曲线的响应均为线性(1/X加权)。
杂质A的标准曲线图示例见图3。尽管进样了高浓度的API,该方法在整个分析过程中,在后续的任何空白进样中均未表现出残留。对于所有浓度≤75 µg/mL的阿托伐他汀API的进样结果都是如此。
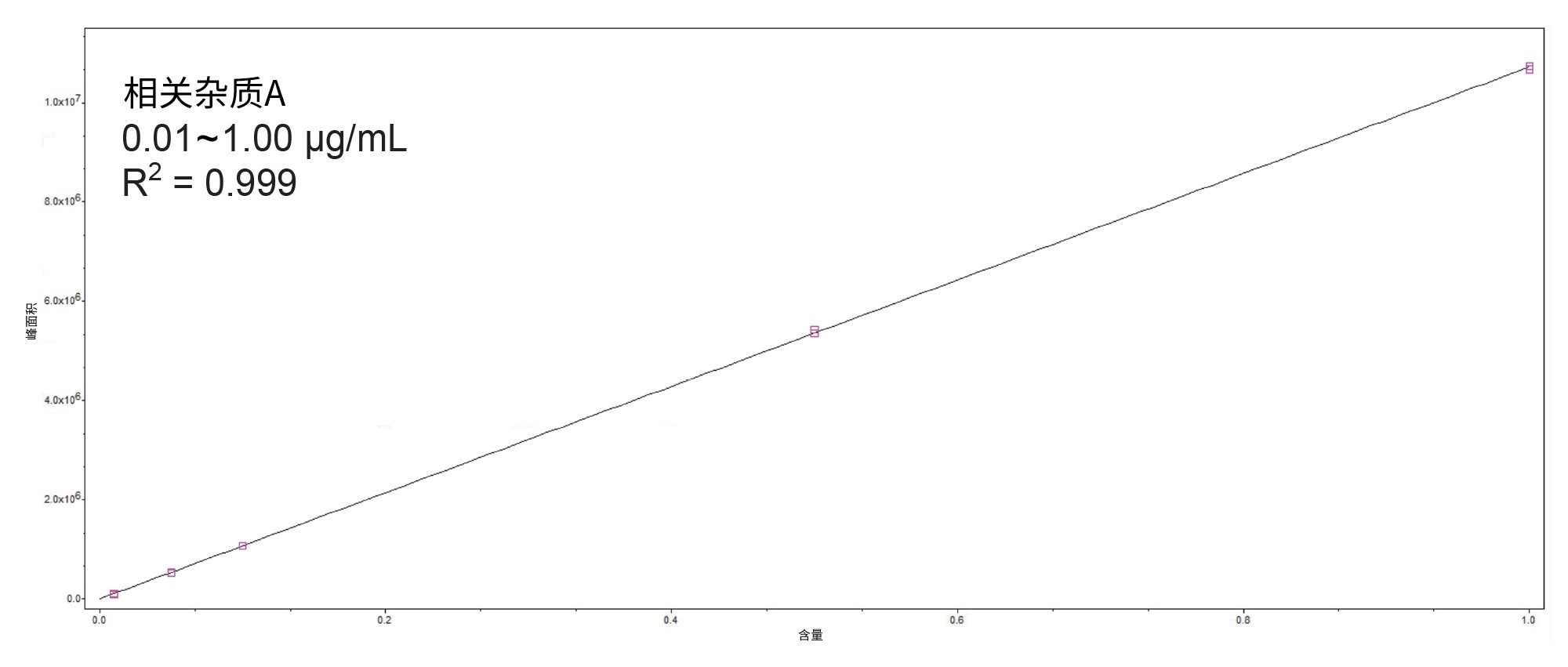
0.15%阈值时阿托伐他汀杂质的定量分析
完成QDa II质谱检测器对相关杂质的灵敏度评估后,向75 µg/mL的阿托伐他汀样品中加标0.12%的杂质混合物(A、C、I,每种杂质的浓度为0.09 µg/mL),进行评估。为简便起见,下文将该样品称为“0.12%阈值样品”。在扫描模式下运用UV和质谱检测技术分析化合物。图4展示了该样品的示例UV色谱图及相关的MS TIC色谱图。
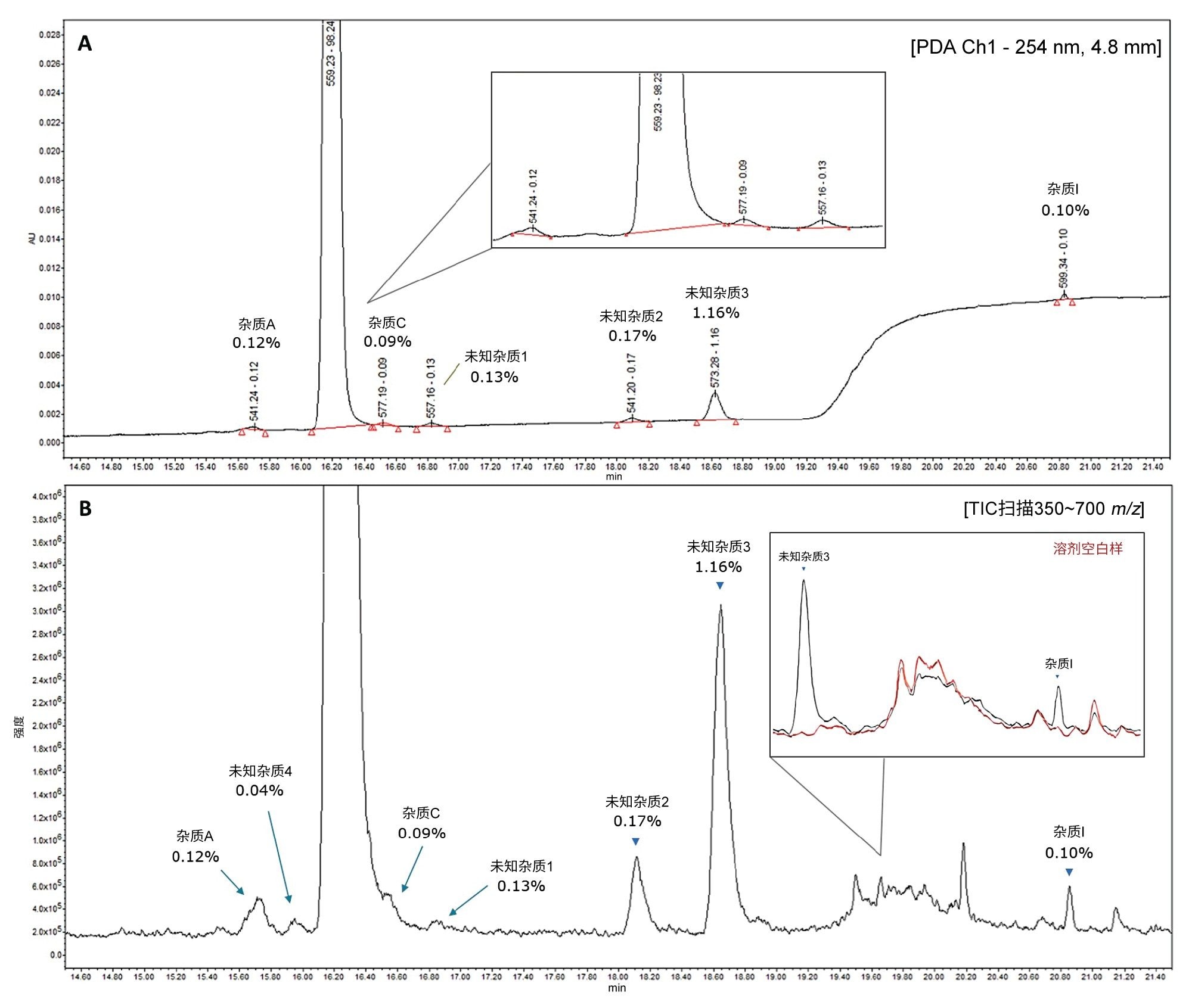
在图4A的PDA迹线中,可以看到三个未知峰,分别为0.13%、0.17%和1.16%,其相对浓度与图1所示25 µg/mL样品中的结果相当。图4B中相应的MS TIC色谱图更清楚地展示了还存在另一种未知杂质,即未知杂质4。
图4B还展示了在同一分析过程中采集的溶剂空白样品的叠加图,表明图4B中的MS TIC色谱图中的未标记峰并非API样品所独有。
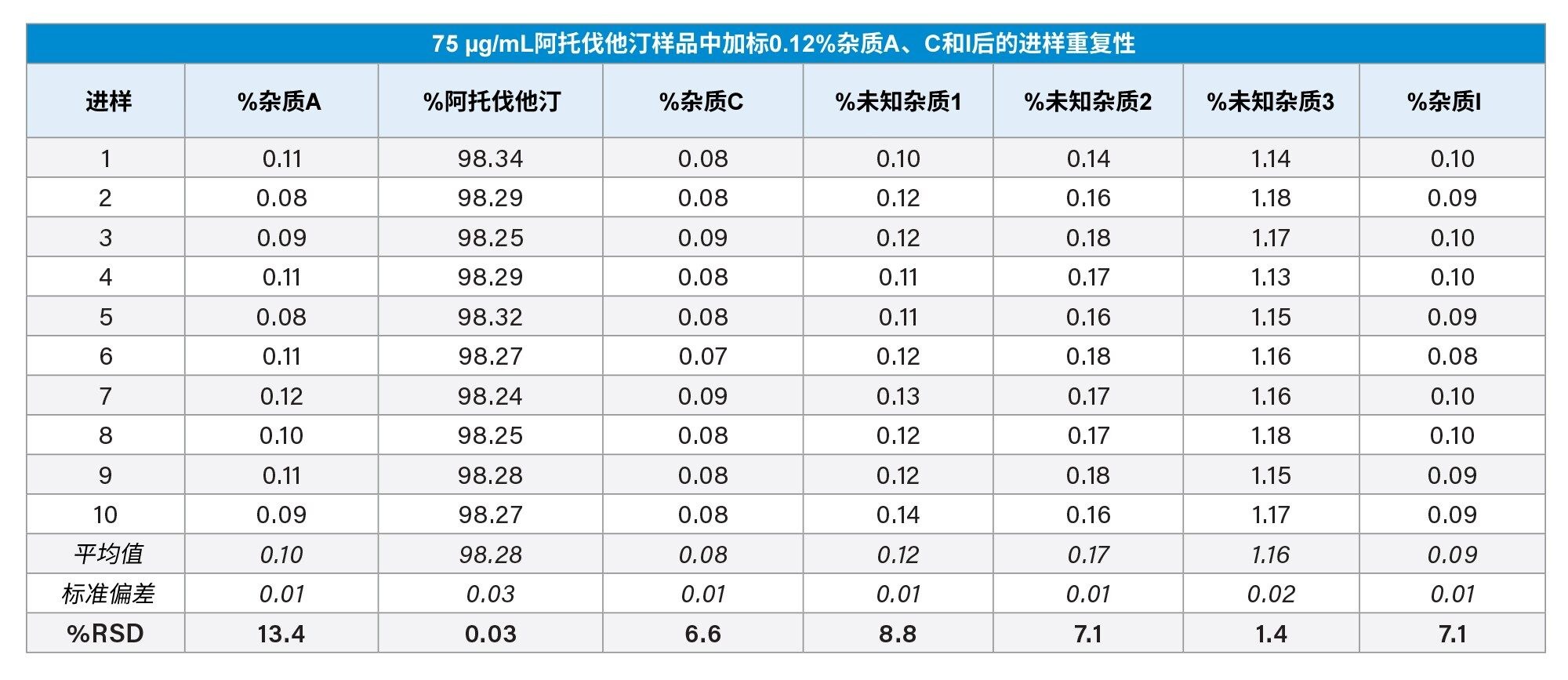
为评估该方法的重复性,0.12%阈值样品进行了重复10次进样。结果请参见表2。除相关杂质A在10次进样中的峰面积%RSD为13.4%以外,其余所有杂质的峰面积%RSD均小于9%。
使用切换阀可以在规定的时间段内自动将液流切换至废液,在MS TIC色谱图中可以更清楚地看到杂质A和C。此功能的使用示例可参见图5,其中液流在16.08 min至16.47 min(阿托伐他汀API洗脱的RT区间)期间切换。
![使用切换阀的MS TIC色谱图,切换阀在API洗脱时将液流引流至废液,从而更轻松地定量杂质A [m/z 541.1]和杂质C [m/z 557.1]](/content/dam/waters/zh/app-notes/2024/720008282/720008282en-f5.jpg.82.resize/img.jpg)
采用ACQUITY QDa II系统,可轻松获取色谱图中每个峰的质谱图(图6)。
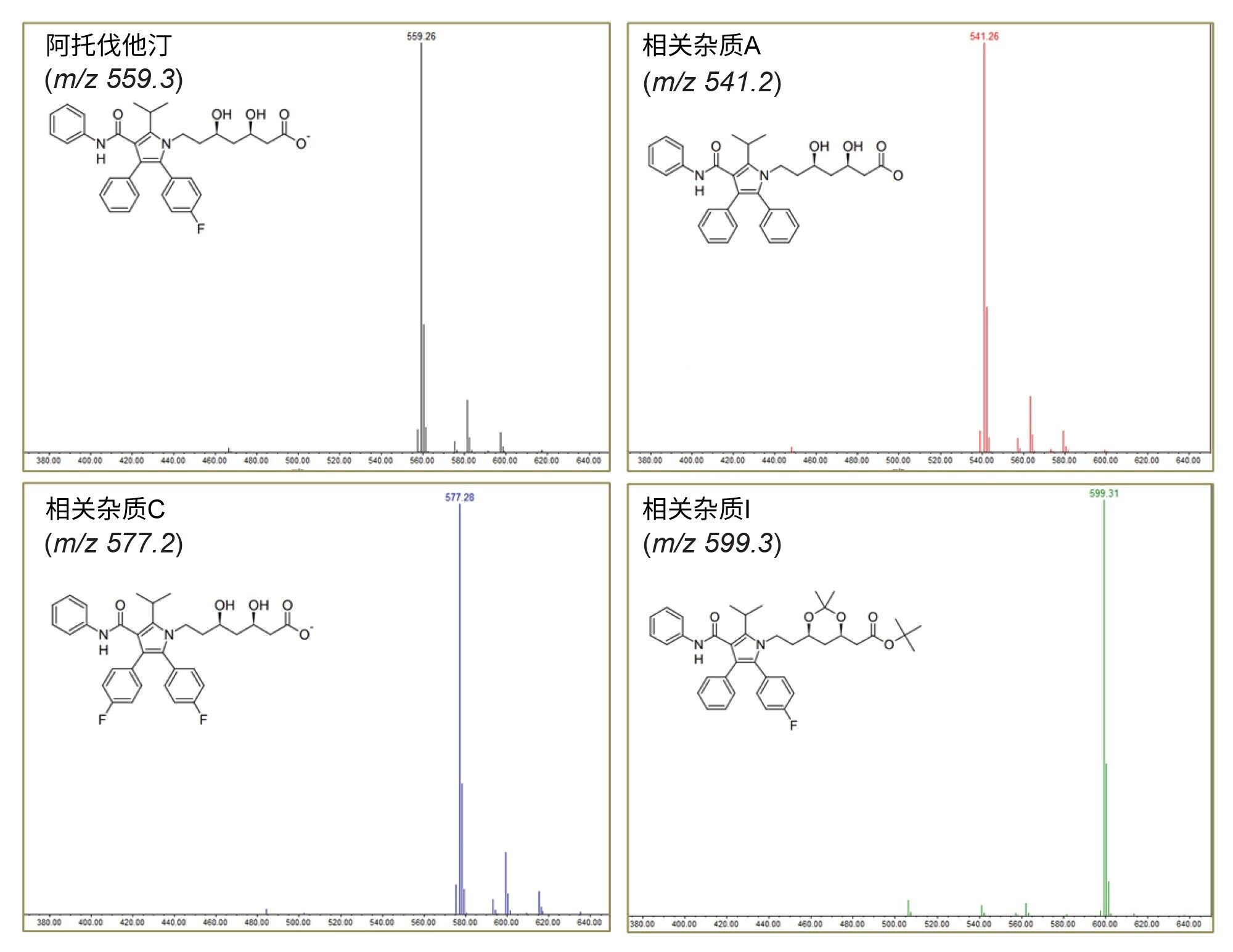
利用源内裂解获取结构信息
研究发现,未知杂质1和4的分子量为m/z 557,仅比API低2 Da。据文献报道,这很可能是一种阿托伐他汀化合物的常见光降解物6。但是,未知杂质2和3需要进一步评估,以便更好地了解其各自的结构。
源内裂解可提供有关分析物分子的重要结构信息,从而检测到有助于鉴定和表征化合物的特征碎片离子。
通过提高QDa II质谱检测器的锥孔电压,可以通过实现源内裂解为给定化合物执行伪MS/MS分析。图7展示了一个研究未知杂质2和杂质3的相关示例。
施加35 V的锥孔电压后,未知杂质3的母离子质量数m/z 573.3碎裂为m/z 454(图7B)。碎片离子谱图表明,该化合物可能是杂质G或阿托伐他汀甲酯。
在该分析中,可以看到m/z 611处有一个加合离子[M+K]+,它在锥孔电压为50 V时进一步碎裂,生成分子量为m/z 436、394、376和292的子离子,该结果与文献中阿托伐他汀甲酯的评估结果更为一致7。然而,要得出绝对结论仍需进行更多分析。
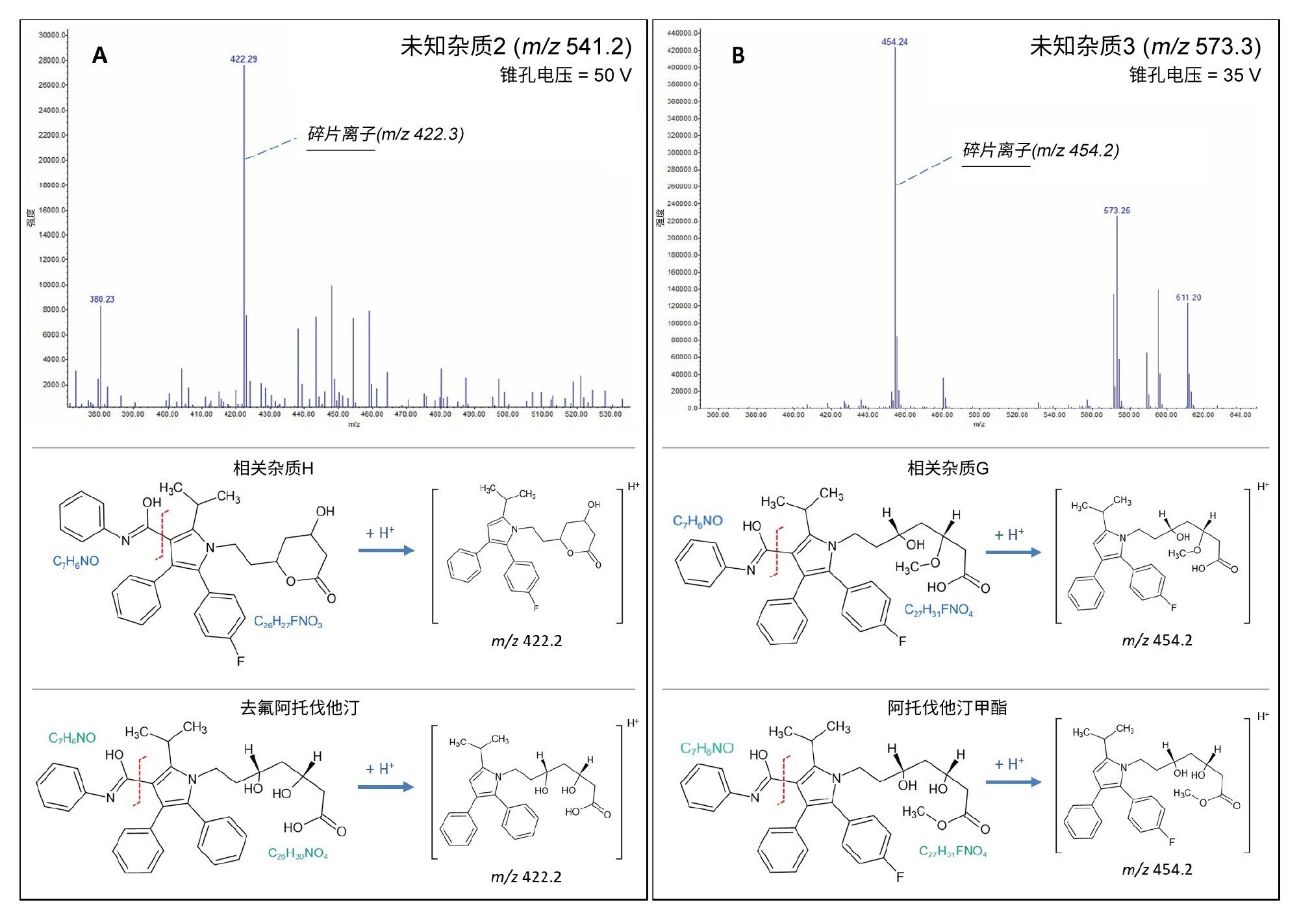
对未知杂质2的m/z 451.2母离子施加50 V锥孔电压,在m/z 422处产生了碎片离子(图7A)。该碎片离子谱图与去氟阿托伐他汀和相关杂质H均匹配。如果后续存在m/z 422的m/z 380碎片离子,则该化合物为相关杂质H的可能性更高。
质谱检测可检出共流出杂质
样品在10 °C下储存两周,以监测杂质随时间的变化。将加标0.12%杂质混合物的75 µg/mL阿托伐他汀样品指定为“降解测试样品”。图8所示的分析结果表明,降解测试样品中相关杂质A的质谱图(图8B)与不含API的相似浓度样品(0.10 µg/mL杂质混合物,图8A)中相关杂质A的质谱图之间存在有趣的差异。在前者中观察到m/z 557.2处显著增加,表明存在API的光降解物并与相关杂质A共流出。
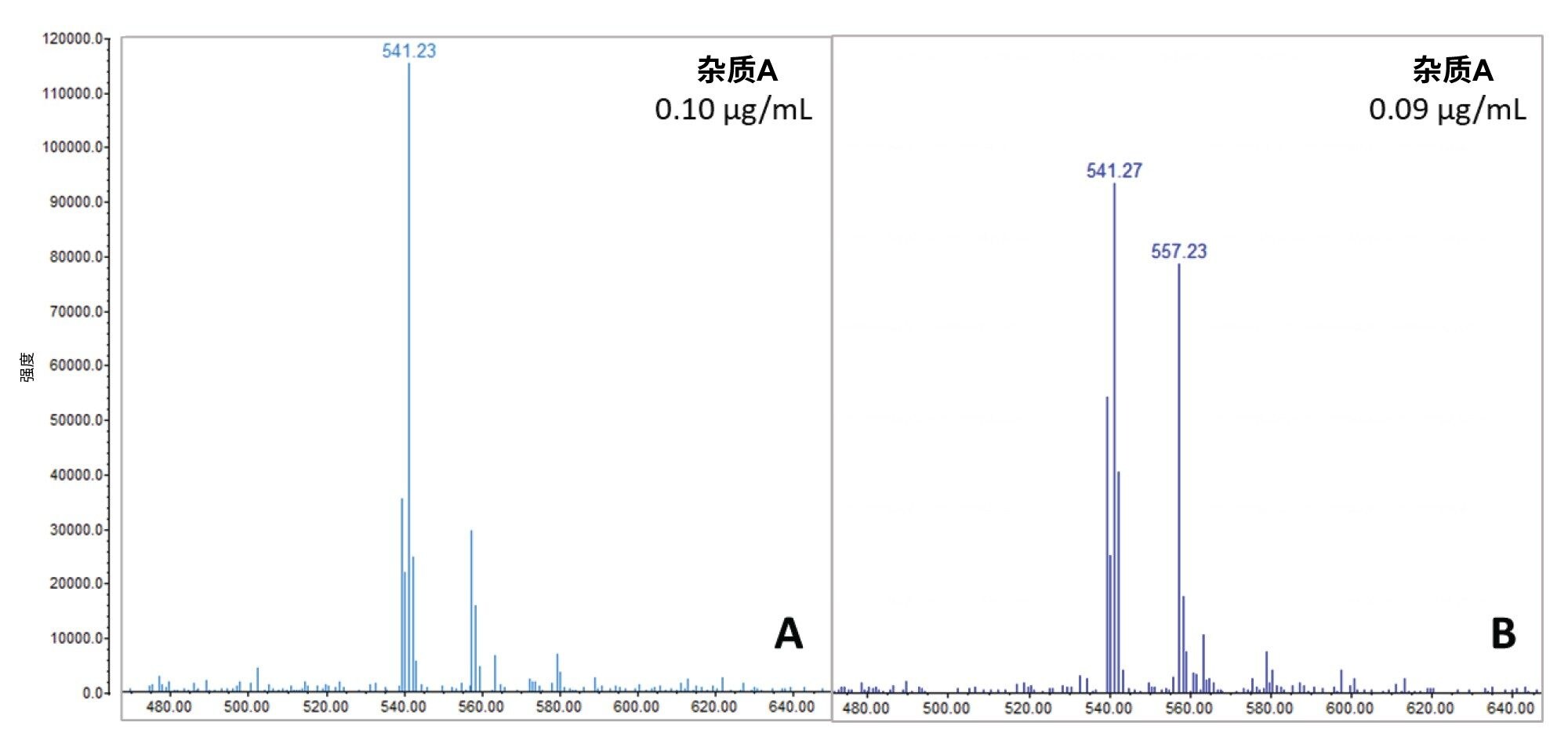
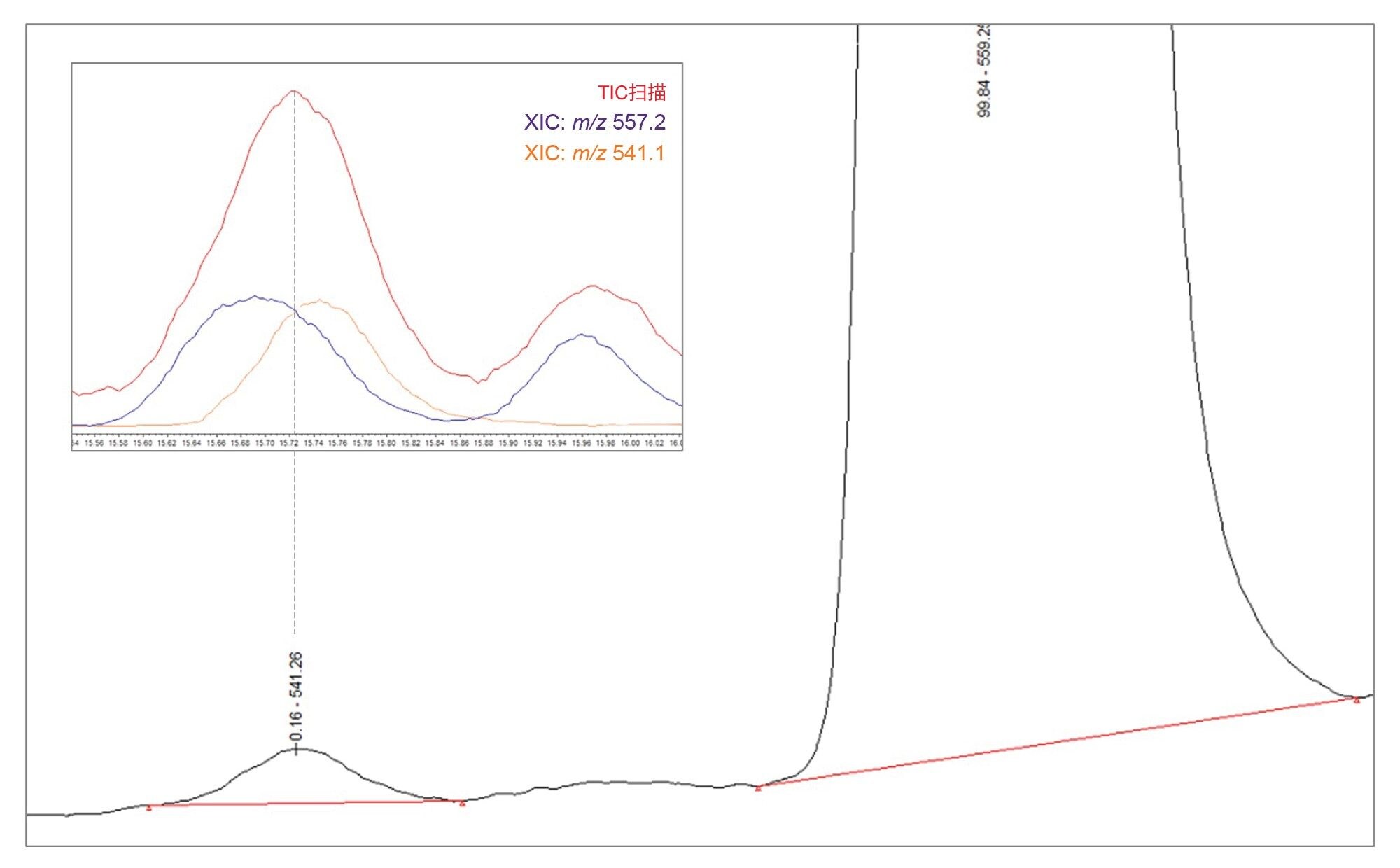
在制备后约15天对样品重新定量时,相关杂质A相对于API的相对含量有所增加,从平均0.10%(见表2)增加到0.16%,如下图9所示。这种差异并非由于相关杂质A的浓度变化,而是由于与另一种化合物的共洗脱,这种现象随着API的降解而变得更加明显。因此,在使用UV色谱进行定量分析时,会导致结果被高估。图9中展示了这一共洗脱现象,其中相关杂质A (m/z 541.1)的XIC与m/z 557.2杂质的XIC以及MS TIC扫描图相叠加。这凸显了质谱检测在确保杂质的准确定量和正确处理共洗脱方面的重要性。
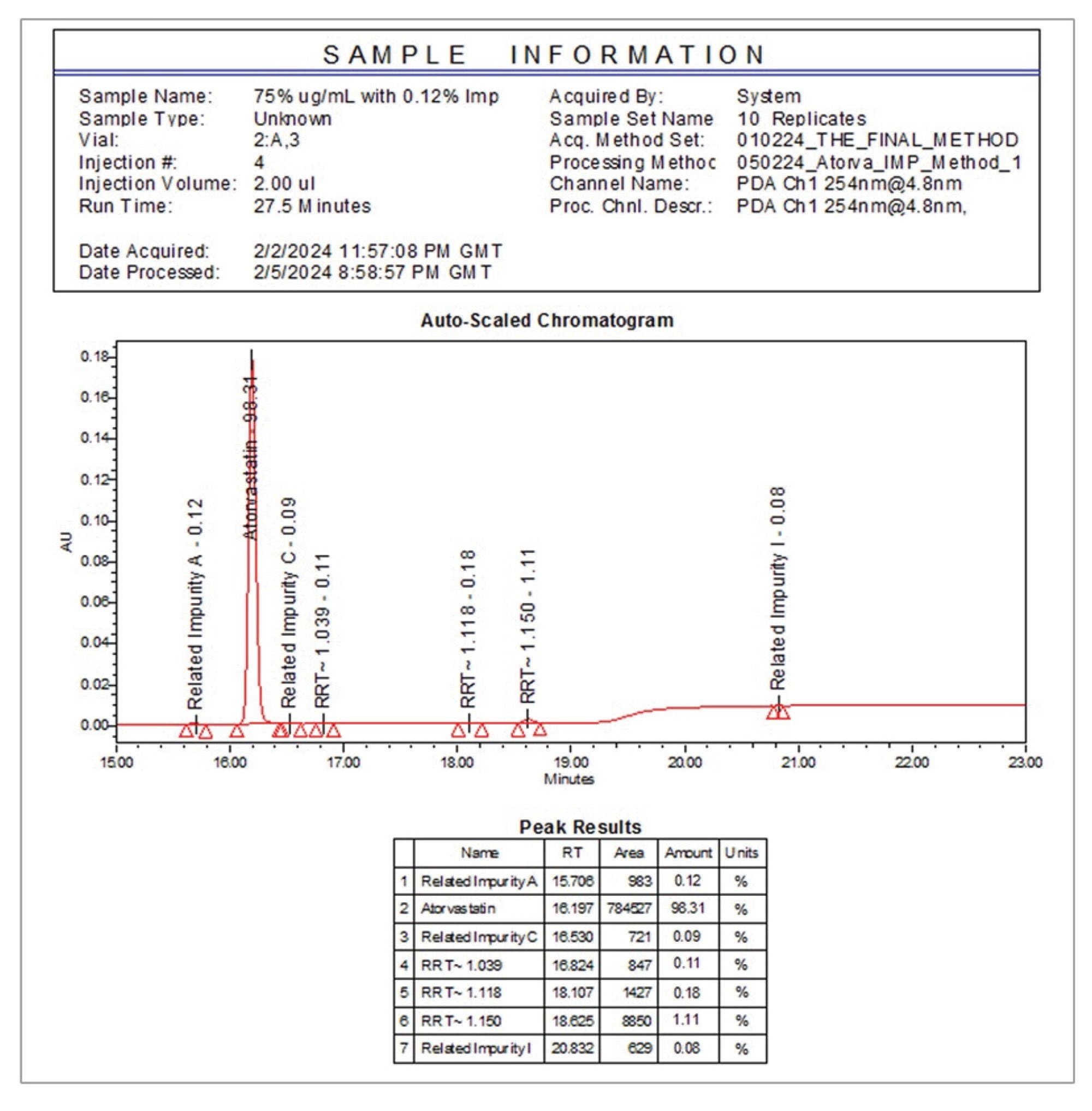
Empower数据报告
本应用广泛利用了Empower CDS报告功能来简化样品批次中相对杂质浓度的评估,如图10所示。这一强大功能可以帮助用户迅速做出明智决策,从而确保工作流程的合规性和数据可靠性。
结论
本研究展示了将ACQUITY QDa II质谱检测器引入现有的UV工作流程中进行API杂质常规检测的显著优势:
- 通过分子量标注峰,在分析中轻松完成质量数确认,简化已知杂质的确认过程
- 精确定量杂质浓度,确保阿托伐他汀相关杂质A、C和I保持在API相对含量0.15%的合格阈值以下
- 通过源内裂解增强结构解析,有助于未知杂质的分子量确认,从而更深入地了解化合物结构
- 借助质谱检测提供的宝贵见解,避免已知杂质报告中的偏差,检测干扰因素、保障产品质量并加快产品放行
结合Empower CDS软件,该分析方法可进一步改善杂质工作流程,实现了简便、高效的解决方案,并符合合规性和数据可靠性标准。
参考资料
- Vukkum P, Moses Babu J, Muralikrishna R. Stress Degradation Behavior of Atorvastatin Calcium and Development of a Suitable Stability-Indicating LC Method for the Determination of Atorvastatin, its Related Impurities, and its Degradation Products.Sci Pharm.Jan-Mar;81(1):93–114.2013.
- Impurities in EW drug substances Q3A(R2)–ICH (Oct 2006) ICH–Q3 Guidelines.请访问:https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf (Accessed: 09 February 2024).
- European Pharmacopoeia (Ph.Eur.)11th Edition.请访问:https://www.edqm.eu/en/european-pharmacopoeia-ph.-eur.-11th-edition (Accessed: 09 February 2024).
- Shulyak, N. et al. ‘Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and Its Impurities in Tablets’, Scientia Pharmaceutica, 89(2), p.16. 2021.
- Piponski, M. et al.‘Concepts in Development of Fast, Simple, Stability Indicating HPLC Method for Analysis of Atorvastatin Related Compounds in Tablets’, Journal of Analytical & Pharmaceutical Research, 7(4), pp.450–457.2018.
- Stach, J. et al. ‘Synthesis of Some Impurities and/or Degradation Products of Atorvastatin’, Collection of Czechoslovak Chemical Communications, 73(2), pp.229-246.2008.
- Mornar, A., Damić, M. and Nigović, B. ‘Separation, Characterization, and Quantification of Atorvastatin and Related Impurities by Liquid Chromatography-Electrospray Ionization Mass Spectrometry’, Analytical Letters, 43(18), pp.2859-2871.2010.
720008282ZH,2024年4月