使用Confirm Sequence结合靶向和非数据依赖型碎片离子数据快速、高效、简便地确认合成寡核苷酸的序列
摘要
本应用纪要展示了使用waters_connect™ CONFIRM Sequence应用程序软件进行合成寡核苷酸的序列确认,相较于传统手动方法,无论是在QTOF(Vion™质谱仪)MS/MS,还是在台式TOF(BioAccord™质谱仪)MSE (DIA)分析中,该软件均展现出了效率的提升。
优势
简介
近年来,合成寡核苷酸开始作为小分子及治疗性蛋白药物的有力补充1。 随着越来越多的寡核苷酸药物进入市场,需要能够提供可靠、稳定质量控制的分析方法2,3。 市售的合成寡核苷酸主要有反义寡核苷酸(ASO)和沉默RNA (siRNA)两大类4,5。 这两种形式通常都采用长度小于50个单体的寡核苷酸。
质谱分析法已被证明是表征此类分子的黄金标准方法,尤其是在使用了经过修饰的核苷和骨架时6-8。 靶向MS/MS碎裂是表征合成寡核苷酸序列的首选方法,这种方法的碎裂途径已得到充分了解和注释9。 近期对寡核苷酸大小、母离子电荷态和碰撞能量之间关系的研究能够帮助我们找到理想条件,生成监管提交所需的序列确认结果10。
该工作流程有两个主要限制因素,一是手动处理所得的复杂数据集,二是发现和早期开发通常使用的质谱仪需要经验丰富的分析人员生成高质量MS/MS碎片离子。这两个局限性都限制了基于MS/MS的序列确认在生产和质量控制实验室的下游应用。
本文展示了使用waters_connect™ CONFIRM Sequence应用程序软件快速获取CpG7909(一种经过修饰的合成寡核苷酸治疗药物)的高质量序列确认数据11。 本文还进一步展示了专为MS技术经验有限的用户设计的台式BioAccord™系统(UPLC™-TOF-MS)在合成寡核苷酸生产常规质量控制方面的潜力。这些结果与寡核苷酸表征常用的QTOF MS(Vion™ MS仪器,通常用于寡核苷酸特性表征)获得的MS/MS数据进行了比较。
手动预测合成寡核苷酸的预期碎片离子颇具挑战。CONFIRM Sequence应用程序中的数据库功能支持用户使用细化至亚组分水平(碱基、糖和连接子)的结构信息创建、管理和存储自定义寡核苷酸序列。应用程序预装的缺省单体条目足以创建CpG7909,但数据库在单体创建和设置自定义碎裂规则方面页也提供了高度的可定制性。
数小时的劳动密集型手动处理可以简化为几分钟的自动化数据处理。借助图形表示,自动标记问题数据区域并高效地管理结果,简化了结果审查。CONFIRM Sequence中使用定制化靶向同位素簇算法,通过直接在原始数据上注释详细的碎片离子匹配,可提供很高的可信度。
实验
样品描述
CpG7909,序列为dT*dC*dG* dT*dC*dG* dT*dT*dT* dT*dG*dT* dC*dG*dT* dT*dT*dT* dG*dT*dC* dG*dT*dT(d代表脱氧核糖核酸,*代表硫代磷酸酯骨架修饰),在BioSprings通过固相合成法内部合成。将CpG7909用水稀释至最终浓度0.5 mg/mL。
液相色谱条件
|
液相色谱系统: |
ACQUITY™ UPLC™二元溶剂管理器 |
|
光学检测: |
TUV |
|
色谱柱: |
ACQUITY UPLC BEH™, 130 Å寡核苷酸分析专用柱, C18 1.7 µm 2.1 × 100 mm |
|
柱温: |
60 °C |
|
进样体积: |
2 µL |
|
流速: |
0.3 mL/min |
|
流动相A: |
含7 mM TEA、80 mM HFIP的水溶液 |
|
流动相B: |
含3.5 mM TEA和40 mM HFIP的50:50甲醇:水溶液 |
|
梯度: |
使用的LC梯度如下:初始条件为10%流动相B缓冲0.2分钟,从0.2分钟至2.0分钟,流动相B从10%增加至100 %,从2.00分钟2.20分钟,保持100%流动相B,从2.20分钟至2.30分钟,从100% B降至10% B,从2.30分钟至3.00分钟保持10% B。柱温60 °C,流速0.3 mL/min。 |
质谱条件
在低质量范围内,以碎裂模式全扫描运行BioAccord™系统RDa™进行50~2000 m/z范围的采集。ESI极性设置为负,扫描速率为1 Hz。将锥孔电压调整为50 V,碎裂锥孔电压从60或70 V开始,逐渐增加至100~120 V。毛细管电压设置为0.8 kV,脱溶剂气温度550 ℃,脱溶剂气流速800 L/h。
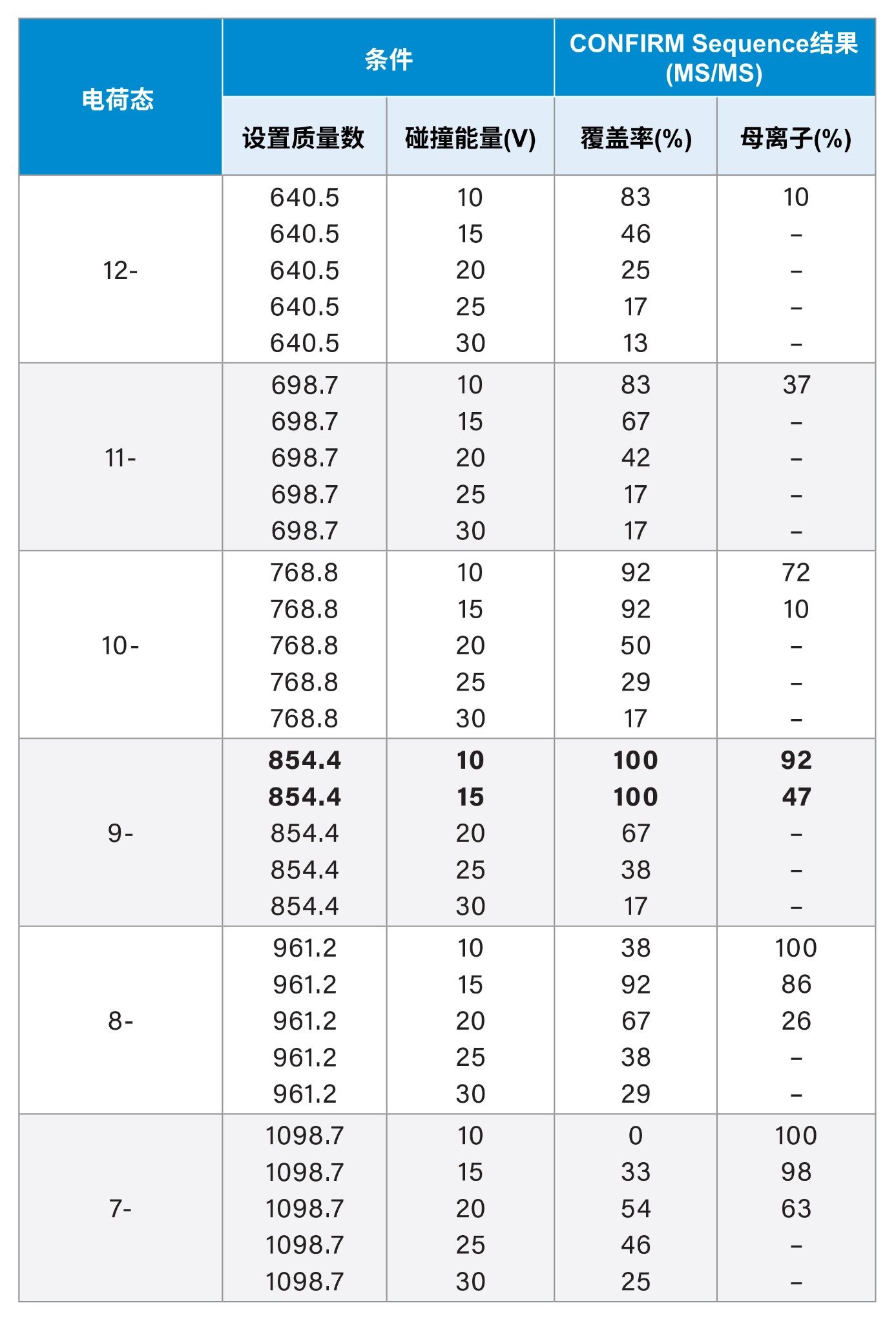
Vion IMS QTOF在灵敏度模式下使用ESI负极性进行标准传输。离子源类型设置为ESI,离子源温度120 ℃,脱溶剂气温度400 ℃,脱溶剂气流速800 L/h,锥孔气流速50 L/h,毛细管电压2.0 kV,样品锥孔电压60 V,离子源补偿电压100V。MS/MS采集的质荷比范围设置为50~2000 m/z,扫描时间为0.5秒,低母离子分辨率。选择的母离子如下(值以m/z表示):640.5(电荷态:12-)、698.7(电荷态:11-)、768.8(电荷态:10-)、854.4(电荷态:9-)、961.2(电荷态:8-)和1098.7(电荷态:7-)。每种母离子的碰撞能量分别在10、15、20、25和30 V之间变化。
处理设置
|
CONFIRM Sequence方法设置(MS/MS) |
|
|
PPM误差: |
20 |
|
强度截止值(计数): |
20 |
|
同位素相似性阈值: |
65 |
|
同位素强度阈值: |
65 |
|
CONFIRM Sequence方法设置(MSe) |
|
|
PPM误差: |
30 |
|
强度截止值(计数): |
200 |
|
同位素相似性阈值: |
65 |
|
同位素强度阈值: |
55 |
结果与讨论
使用QTOF MS进行寡核苷酸测序涉及电荷态选择和碰撞能量筛选,以获得理想结果。根据电荷态的不同,需要优化碰撞能量,避免低能量的部分碎裂和高能量的过度碎裂(序列的内部断裂和碎片离子碎裂)。
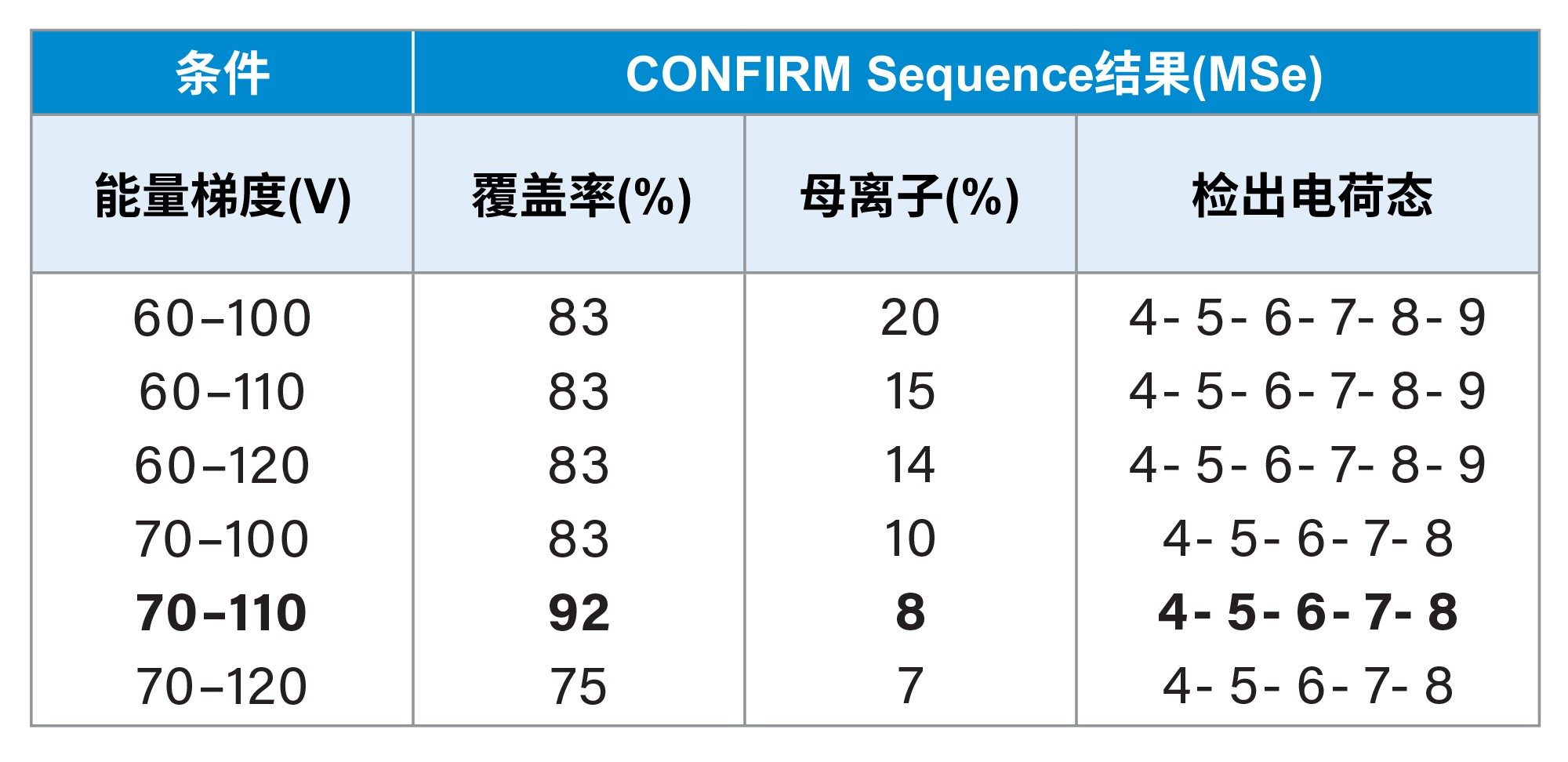
对于我们的测试分子CpG7909(一种免疫刺激性佐剂合成寡核苷酸治疗药物),Gawlig和Rühl(2023年)对低电荷态所需增加能量的观察,在MS/MS(表1)和MSe(表2)分析中也有所体现10。
另一种针对碎片离子的调谐策略为,以存在于碎片离子数据中的残留母离子量为靶向。如果不存在残留的母离子信号,则分子可能过度碎裂,或者在给定条件下电荷态不稳定。在所有情况下,CONFIRM Sequence中显示的母离子百分比值的降低都与碰撞能量增加有关,并且还可能表明碰撞能量增加导致了过度碎裂。总体而言,对于给定的电荷态,存在母离子情况下的最高碰撞能量即可指示最佳设置,并且可以使用%母离子指标来获得该优化结果。
此处的数据显示,进行MSe碎裂时,能够产生少量残留母离子(5%~10%)的最高碰撞能量较为理想。而对于MS/MS碎裂,理想条件取决于电荷态。对于相同的电荷态,与较低能量的碰撞能量相比,如果序列覆盖率和母离子百分比降低,则表明存在过度碎裂。优化MS/MS采集条件时,应尝试增加碰撞能量,直至母离子百分比低于60%,然后继续增加能量,直到获得与较低能量相比可以增加序列覆盖率的最低母离子百分比。
手动处理与CONFIRM Sequence
通过手动工作流程审查数据非常耗时,且容易发生用户错误。CONFIRM Sequence中的审查步骤旨在以简单易懂的方式展示序列覆盖率,并提供相关功能,让用户能够手动审查直接在原始数据上注释的每个碎片离子匹配。
CONFIRM Sequence中的数据处理速度非常快(不到5分钟即可处理完此处展示的所有数据),并且可由专家用户进行预配置,让经验不足的用户也能在专家指导下处理大型数据集。此外,waters_connect™软件平台符合法规要求,包括质量保证实验室运营中所需的报告、数据可靠性控制和方法版本管理。
CONFIRM Sequence应用程序提供高保真度碎片离子匹配,并且从覆盖率计算中移除了同分异构体碎片分配,从而减少了假阳性分配,减轻了用户手动数据确证的负担。
提高匹配保真度
经过修饰的合成寡核苷酸在较大质量范围内的复杂碎片离子数据可能需要相当长的去卷积时间(每幅谱图耗时长达20分钟),并且需要一定的专业知识才能手动研究去卷积结果并根据质量数分配预测的碎片离子。
CONFIRM Sequence应用程序采用智能靶向同位素聚类算法,能够解决手动非靶向分析方法所面临的挑战。CONFIRM Sequence以靶向方式处理数据,直接针对每个碎片离子的预测同位素簇搜索原始数据,并基于多种评分指标进行匹配。用户可以在应用程序中查看原始数据的匹配结果,为碎片离子分配提供支持。
此外,系统还应用了一组逻辑规则来控制如何使用碎片离子组来确认单体分配。其中的一个例子是使用碱基丢失的碎片离子(例如,a-b-离子),离子在碰撞诱导解离碎裂后经历气相离子/离子重排,导致碱基丢失。由于存在这种丢失,仅使用碱基丢失离子来确认是否存在直接位于碎裂位点5'端的单体是不合理的。CONFIRM Sequence中的逻辑规定,这类离子应该用于n-1系列的碎片离子(例如,a-b3离子不应用来确认第三个单体的存在,而应在[x-]2离子系列中使用)。CONFIRM Sequence中使用的这种分配逻辑旨在确保在应用程序中确认的单体是基于可预测的碎裂建模和可观测的数据。软件中提供有帮助文件,其中更详细地解释了每条单体确认规则。
不确定性
CpG7909具有多个预测的同分异构体碎片离子,使用它们来确认序列的多个部分将是不真实的。CONFIRM Sequence会标记这些不确定的序列,并将其从初始序列确认组中删除,但提供用户在查看数据时将其添加回序列中的选项。例如,我们重新添加回w1-离子,由于5'端和3'端单体相同,w1-与d1-为同分异构体。在本例中,这些单体均位于序列末端,因此我们认为允许这种模糊匹配是可以接受的。然而,CpG7909序列中存在多个同分异构体碎片离子,无法合理地用来确认序列,因此难以覆盖序列的某些部分。而这确实凸显出执行手动序列确认工作的风险,因为这些不确定序列不容易被识别。
比较靶向MS/MS与数据非依赖型采集(MSe)
CONFIRM Sequence能够在单幅MS/MS碎片离子谱图中展示出100%的序列覆盖率,在BioAccord系统上使用数据非依赖型碎片离子模式采集(DIA)时的序列覆盖率为92%。MSe (DIA)采集的优势在于其简化的设置,能够通过一个碰撞能量梯度碎裂所有母离子,该梯度设计用于匹配需要使用靶向MS/MS采集而提前确定的优化CE设置。因此用户无需预先选择目标母离子电荷态和优化碰撞能量的实验设计。
为了协助优化MS/MS实验,CONFIRM Sequence应用程序将每次MS/MS采集的结果同时显示在一个概览表中(图1),其中显示了合并序列覆盖率以及一个展示每幅谱图单独相关信息的表格。用户可以根据所有数据立即确定理想碎裂条件,并深入了解哪种电荷态需要更多/更少的能量才能达到理想条件。
在CONFIRM Sequence应用程序中查看单个分析碎裂结果的流程非常简单,每个碎片离子分配均提供带注释的谱图,用户可以浏览谱图并在必要时剔除(图2)。系统会同时标记出不确定的碎片离子分配结果以供查看。
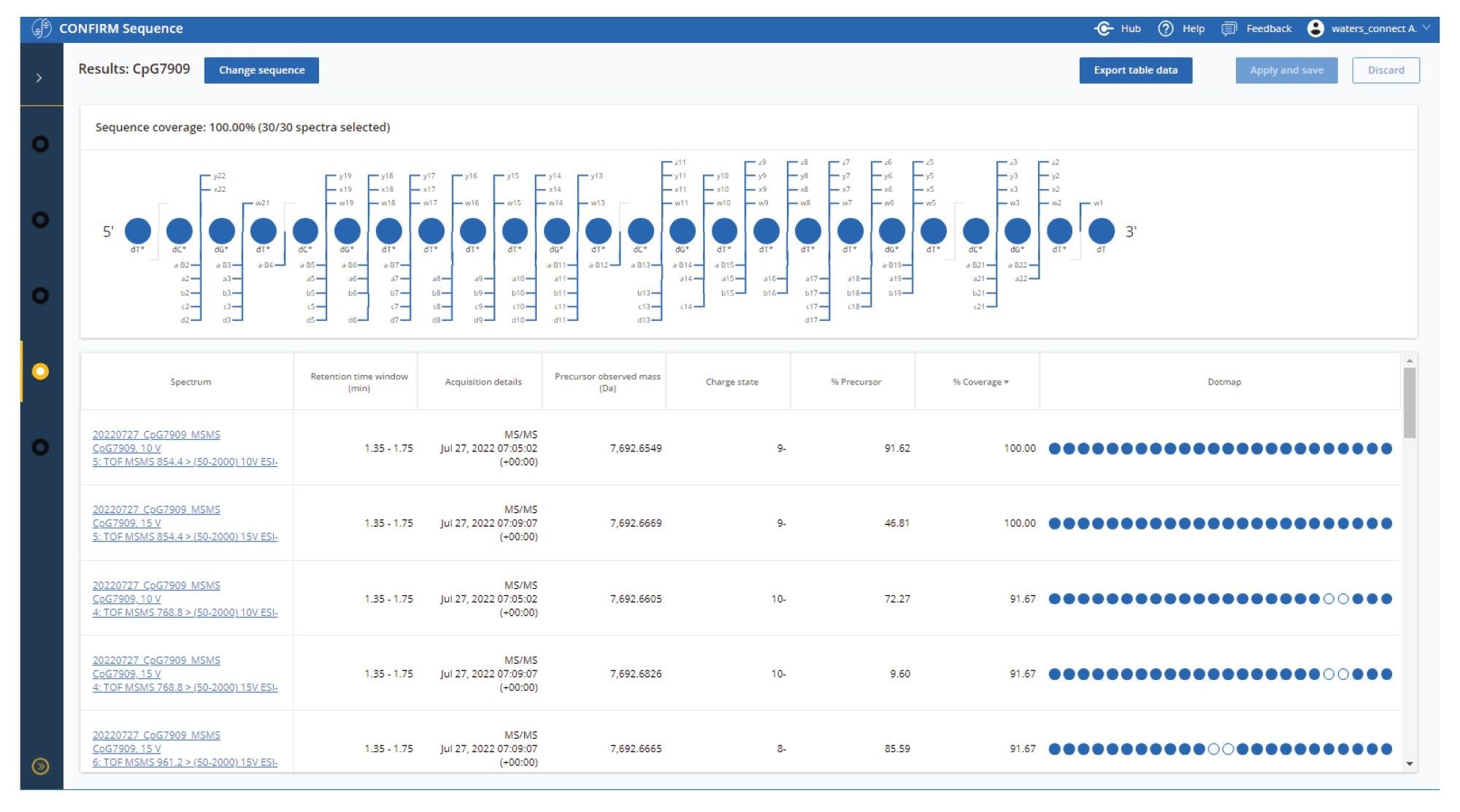
图1.展示结果的CONFIRM Sequence应用程序结果概览页面屏幕截图。此处所示数据为CpG7909的MS/MS实验结果,目标母离子(m/z) 640.5 (-12)、698.7 (-11)、768.8 (-10)、854.4 (-9)、961.2 (-8)和1098.7 (-7)。每种母离子的碰撞能量分别在10、15、20、25和30 V之间变化。Dotmap™视图中显示了合并序列覆盖率的概览信息,其中序列以彩色球和碎片离子连接线表示,表明基于在原始数据中匹配到的碎片离子,单体得到确认(蓝球)。未确认的单体用白色球表示。
该表展示了各个MS/MS谱图的结果,包括以下信息:进样/通道名称、保留时间、采集详细信息、观测到的母离子质量数、鉴定出的母离子电荷态、检测到的残留%母离子和%覆盖率,并用简化的Dotmap™展示了已确认的单体。按序列覆盖率对表格进行排序后发现,两种单独的MS/MS条件能够产生100%的序列覆盖率(碰撞能量10V和15V下电荷态-9)。%母离子从10 V时的91.62%降至15 V时的46.81%。当使用20 V时,未检测到该电荷态的母离子,表明在此能量下发生了过度碎裂。
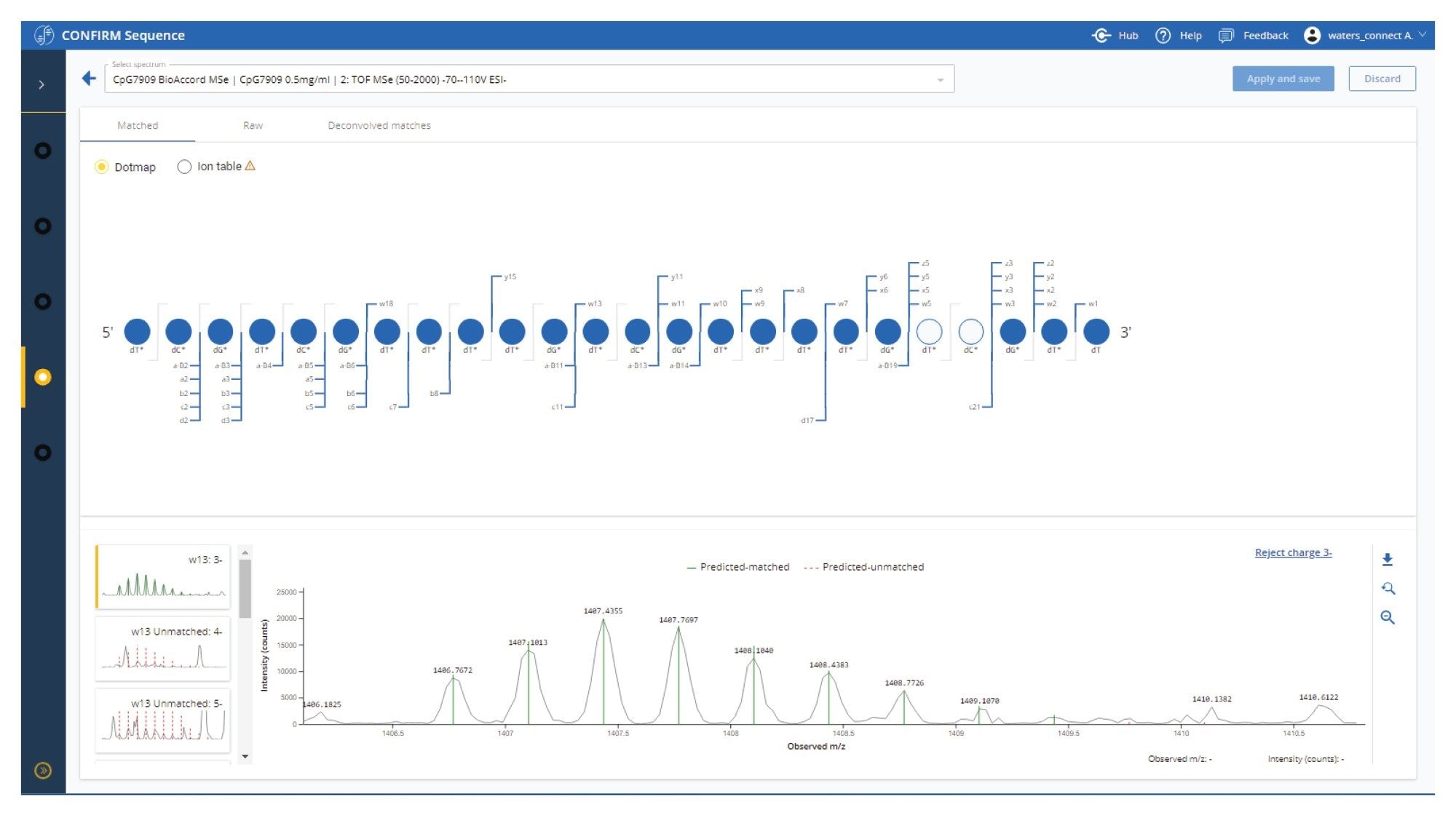
CpG7909,序列为dT*dC*dG* dT*dC*dG* dT*dT*dT* dT*dG*dT* dC*dG*dT* dT*dT*dT* dG*dT*dC* dG*dT*dT(d代表脱氧核糖核酸,*代表硫代磷酸酯骨架修饰)。
结论
CONFIRM Sequence waters_connect™应用程序非常适合用于靶向MS/MS和数据非依赖型碎片离子数据。
合成寡核苷酸序列确认的手动去卷积和碎片离子分配非常耗时,也更容易出现用户错误,并且高度依赖于用户经验。相比之下,自动化的CONFIRM Sequence工作流程产生的结果与手动工作流程相当,但包含了以下优势:自动化碎片离子匹配、直接匹配原始数据、处理速度提高,以及在序列覆盖率结果中考虑同分异构体不确定性。
BioAccord是一款节省空间、经济有效的LC-MS系统,专为非质谱专家设计,基于合规性平台,扩展了自动化数据处理的可用性,适用于受监管的生产和质量实验室。CONFIRM Sequence应用程序能够处理和注释来自BioAccord系统的DIA碎片数据,并通过自动化工作流程实现高可信度的分配。该TOF MS系统获得的优质高能量数据使其成为QTOF或其他高端质谱系统的优选替代选择。
QTOF系统对分子的初始表征具有更好的碎裂控制和极高的灵敏度,但对于此类常规序列确认挑战而言,可能有点大材小用。CONFIRM Sequence应用程序实用性强,对这两种工作流程均提供支持,有助于减轻培训负担,并帮助分子和方法从开发到生产和QC组织的过渡更加平稳。
参考资料
- Sharma, V.K., Watts, J.K.: Oligonucleotide Therapeutics: Chemistry, Delivery, and Clinical Progress.http://dx.doi.org/10.4155/fmc.15.144.7, 2221–2242 (2015).https://doi.org/10.4155/FMC.15.144
- Al Shaer, D., Al Musaimi, O., Albericio, F., de la Torre, B.G.: 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest.Pharmaceuticals.15, (2022).https://doi.org/10.3390/PH15020222
- Musaimi, O. Al, Shaer, D. Al, Albericio, F., de la Torre, B.G.: 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals.14, 1-14 (2021).https://doi.org/10.3390/PH14020145
- Dhuri, K., Bechtold, C., Quijano, E., Pham, H., Gupta, A., Vikram, A., Bahal, R.: Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med. 9, 1–24 (2020).https://doi.org/10.3390/JCM9062004
- Rossi, J.J., Rossi, D.J.: siRNA Drugs: Here to Stay.Molecular Therapy.29, 431-432 (2021).https://doi.org/10.1016/J.YMTHE.2021.01.015
- McLuckey, S.A., Habibi-Goudarzi, S.: Ion Trap Tandem Mass Spectrometry Applied to Small Multiply Charged Oligonucleotides with a Modified Base.J Am Soc Mass Spectrom.5, 740-747 (1994).https://doi.org/10.1016/1044-0305(94)80006-5
- Mcluckey, S.A., Van Berkel, G.J., Glish, G.L.: Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides.J Am Soc Mass Spectrom.3, 60-70 (1992).https://doi.org/10.1016/1044-0305(92)85019-G
- Abdullah, A.M., Sommers, C., Hawes, J., Rodriguez, J.D., Yang, K.: Tandem Mass Spectrometric Sequence Characterization of Synthetic Thymidine-Rich Oligonucleotides.Journal of Mass Spectrometry.57, (2022).https://doi.org/10.1002/JMS.4819
- Wu, J., McLuckey, S.A.: Gas-Phase Fragmentation of Oligonucleotide Ions.Int J Mass Spectrom.237, 197-241 (2004).https://doi.org/10.1016/J.IJMS.2004.06.014
- Gawlig, C., Rühl, M.: Investigation of the Influence of Charge State and Collision Energy on Oligonucleotide Fragmentation by Tandem Mass Spectrometry.Molecules 2023, Vol.28, Page 1169.28, 1169 (2023).https://doi.org/10.3390/MOLECULES28031169
- Cooper, C.L., Davis, H.L., Morris, M.L., Efler, S.M., Al Adhami, M., Krieg, A.M., Cameron, D.W., Heathcote, J.: CPG 7909, An Immunostimulatory TLR9 Agonist Oligodeoxynucleotide, as Adjuvant to Engerix-B® HBV Vaccine in Healthy Adults: A double-blind phase I/II study.J Clin Immunol.24, 693-701 (2004).https://doi.org/10.1007/S10875-004-6244-3/METRICS
致谢
感谢BioSpring GmbH的Alexander Litz协助进行数据生成。
waters_connect、Vion、BioAccord UPLC、ACQUITY和RDa是沃特世公司的商标。
感谢BioSpring GmbH的Hüseyin Aygün为本研究提供的支持。
Michael Rühl和Güngör Hanci就职于BioSpring GmbH(Alt-Fechenheim 34, 60386, 德国法兰克福)。
720008079ZH,2023年11月