在法医毒理学研究中使用简单的“稀释-上样”方法对尿液中的镇痛药物和滥用药物进行UPLC-MS/MS分析
仅适用于法医毒理学应用。
摘要
本应用简报介绍了在法医毒理学研究中使用简单的“稀释-上样”方法进行镇痛药物和滥用药物的UPLC-MS/MS分析,并针对沃特世应用纪要720006187ZH中描述的方法提供了一种替代方案。样品前处理过程从SPE简化到只需一步稀释,与此同时仍能保持极低的残留、一致的基质效应和精确的定量数据,使组内大多数分析物都能达到所需的分析灵敏度。如果化合物对分析灵敏度的要求更高,则建议进一步净化样品,如原始应用纪要所述。
ACQUITY™ UPLC™ BEH™ C18色谱柱可以快速分析大量化合物,同时保持所需的分离度,确保不受同分异构体化合物的干扰。
Waters™ Xevo™ TQ-S micro IVD能在较宽的动态范围内准确定量大量分析物,少至2 ng/mL,多至2500 ng/mL的分析物都能同时定量。

样品萃取、色谱分离与MS/MS检测相结合,得到一套简单的工作流程方法,既快速又精确,还可以在Hamilton STAR或STARlet系统上自动化运行。
优势
- 通过快速、简单的方法分析一组全面的确证药物
- 采用简单的“稀释-上样”样品萃取方法
- 一致的基质效应和回收率,残留极低
- 精确定量大量确证药物
简介
在法医毒理学分析中,分析物组通常包括违禁药物和常见的滥用药物。通常,研究人员使用多种方法获取多种药物的全面信息。这些方法可包括免疫分析、GC-MS、LC-MS/MS或组合方法。沃特世开发了一种用于定量分析综合性药物组的方法,以获得适当的分析灵敏度、选择性和准确度,从而在法医毒理学中实现明确鉴定。
该方法采用“稀释-上样”方法和ACQUITY UPLC BEH C18色谱柱,通过简单的样品萃取方案以及快速、可重现的色谱方法,使所有存在潜在干扰的关键分析物对均能实现基线分离。Waters Xevo TQ-S micro IVD质谱仪具有Xtended Dynamic Range (XDR)功能,能够提供多种化合物所需的分析灵敏度和动态范围。尽管该方法已被证明能够为镇痛药物和滥用药物提供合适的结果,但由于采用的是简化的“稀释-上样”工作流程,因此在分析灵敏度方面也存在一定局限性。如果只有少数分析物对分析灵敏度的要求不高,则建议进一步净化样品,如沃特世应用纪要720006187ZH中所述。
实验
样品萃取
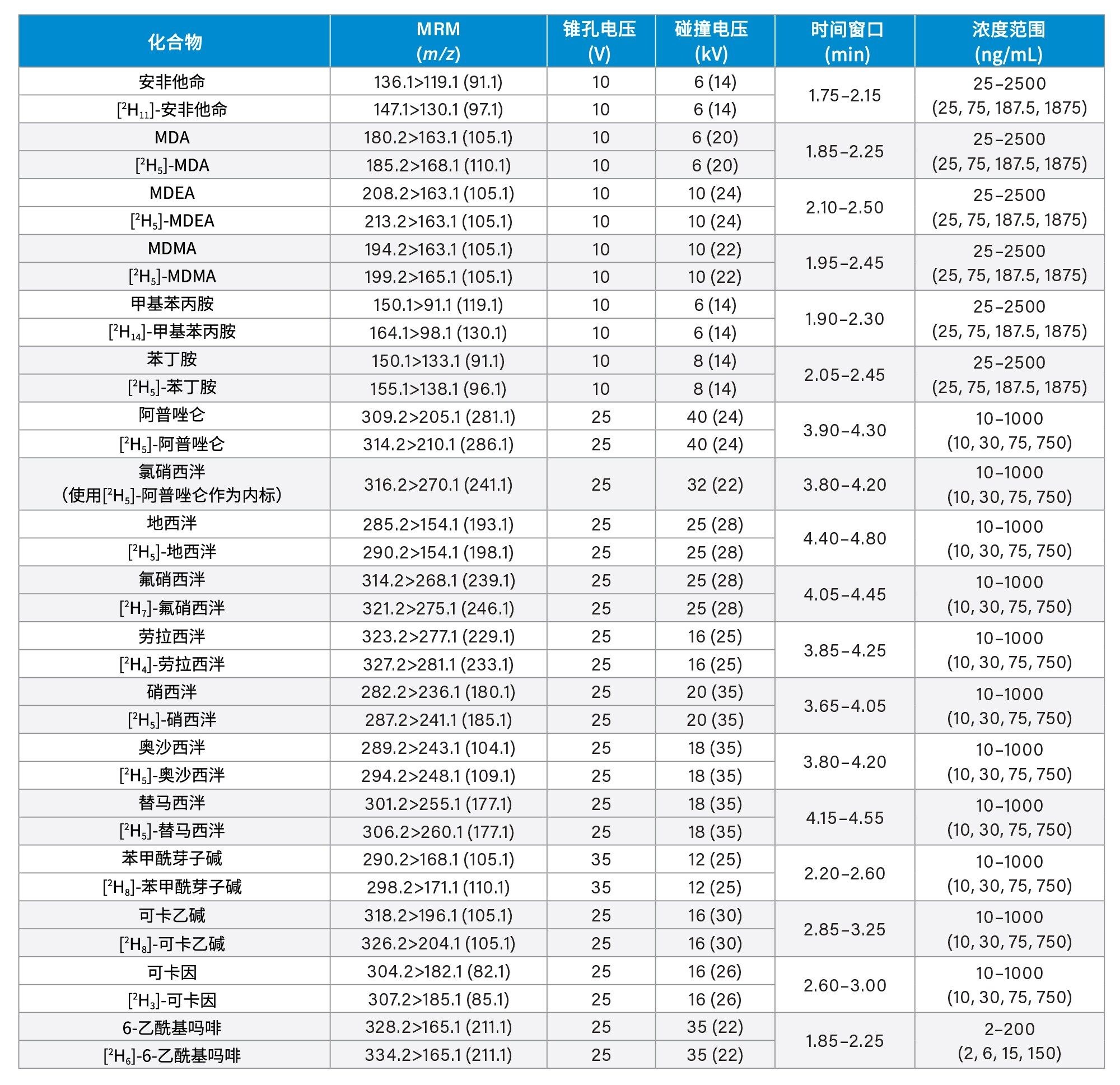
所有标准品均购自Cerilliant(Merck Life Sciences,英国吉林汉姆)、Toronto Research Chemicals(安大略省北约克)和Cambridge Biosciences UK(英国剑桥)。用甲醇配制浓度为2、10和25 µg/mL的混合储备液,具体取决于分析物种类。用50/50 (v/v)甲醇/水配制浓度为100 ng/mL的内标工作溶液。除氯硝西泮、去氢去甲氯胺酮、甲沙酮、去甲羟吗啡酮和α-吡咯烷戊苯酮(α-PVP)代谢物1外(这些化合物不容易获得稳定标记的内标),其余所有化合物均使用了稳定同位素标记的内标。稀释储备液,然后将稀释液添加到混合的空白尿液中,制备标样。同样,稀释储备液,然后将稀释液添加到混合的空白尿液中,制备质控(QC)样品。附录1列出了所有分析物的保留时间和标准曲线范围。
在样品萃取时,可以利用Hamilton液体处理机器人实现半自动化,使样品从样品管条形码到处理样品的每一个环节均可追踪。
取25 μL尿液样品转移至沃特世700 μL圆形96孔收集板中,加入内标工作溶液并充分混合。在进样到UPLC-MS/MS系统之前,用蒸馏水/甲酸溶液稀释并混合样品。
液相色谱条件
|
液相色谱系统: |
ACQUITY UPLC I-ClassFL IVD |
|
色谱柱: |
ACQUITY UPLC BEH C18, 1.7 µm, 2.1 x 100 mm |
|
柱温: |
40 °C ± 2 °C报警 |
|
进样体积: |
20 µL |
|
流动相A: |
0.1%甲酸的水溶液 |
|
流动相B: |
含0.1%甲酸的乙腈溶液 |
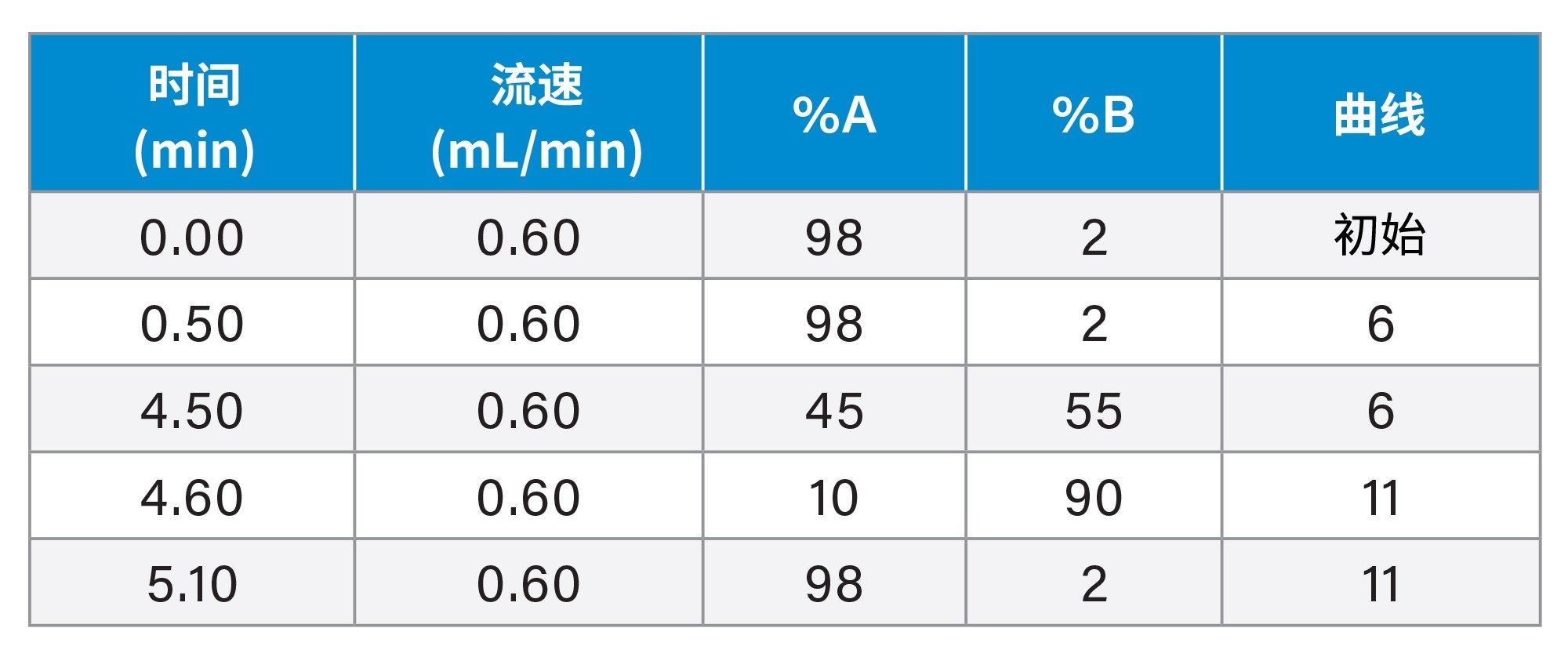
梯度表
质谱条件
|
质谱系统: |
Xevo TQ-S microIVD |
|
电离模式: |
ESI+ |
|
毛细管电压: |
0.8 kV |
锥孔电压、碰撞能量和多重反应监测(MRM)通道等MS方法参数见附录1。
数据管理
|
MS软件: |
MassLynx |
|
信息学软件: |
TargetLynx™ |
结果与讨论
色谱分析
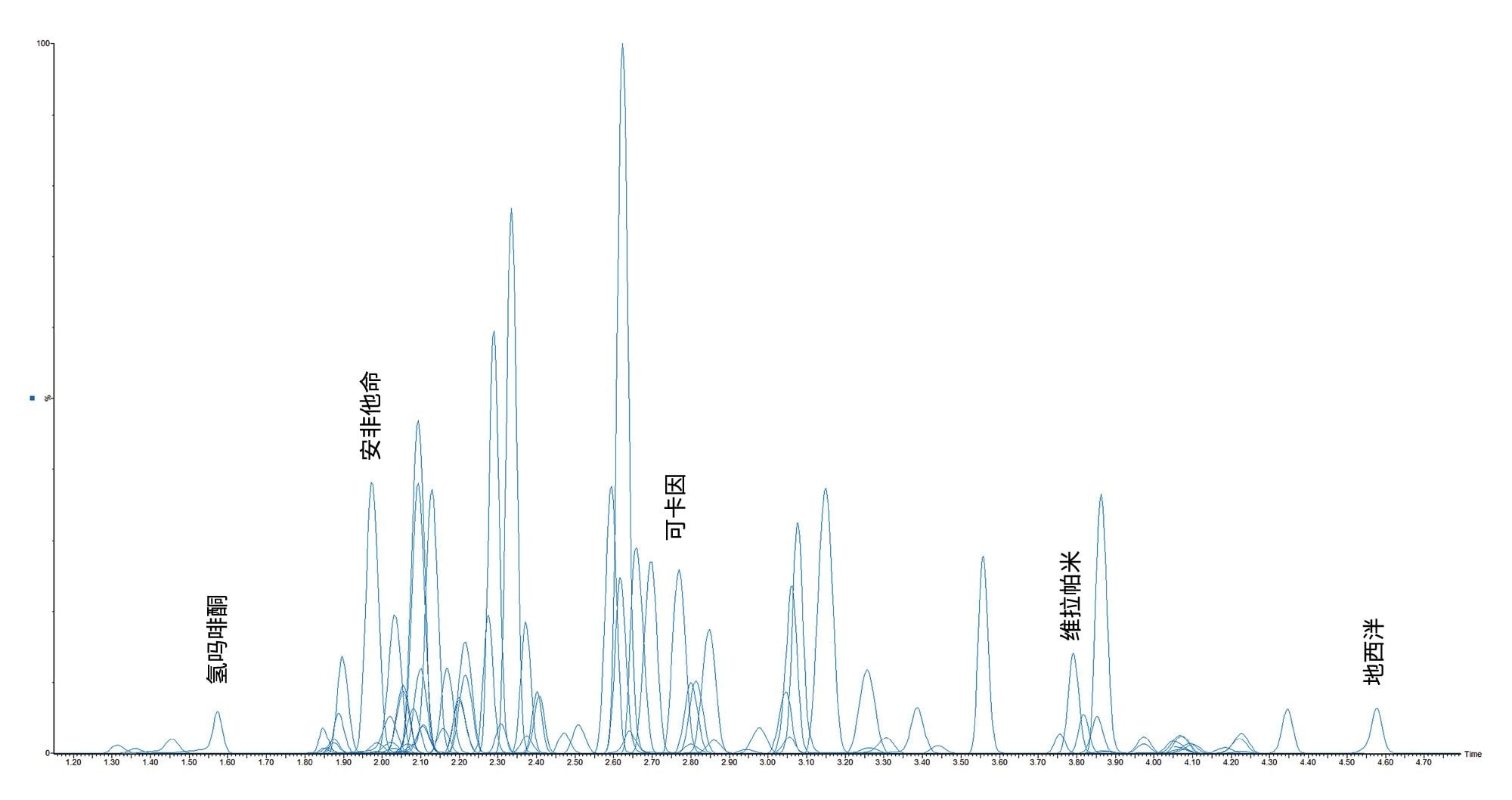
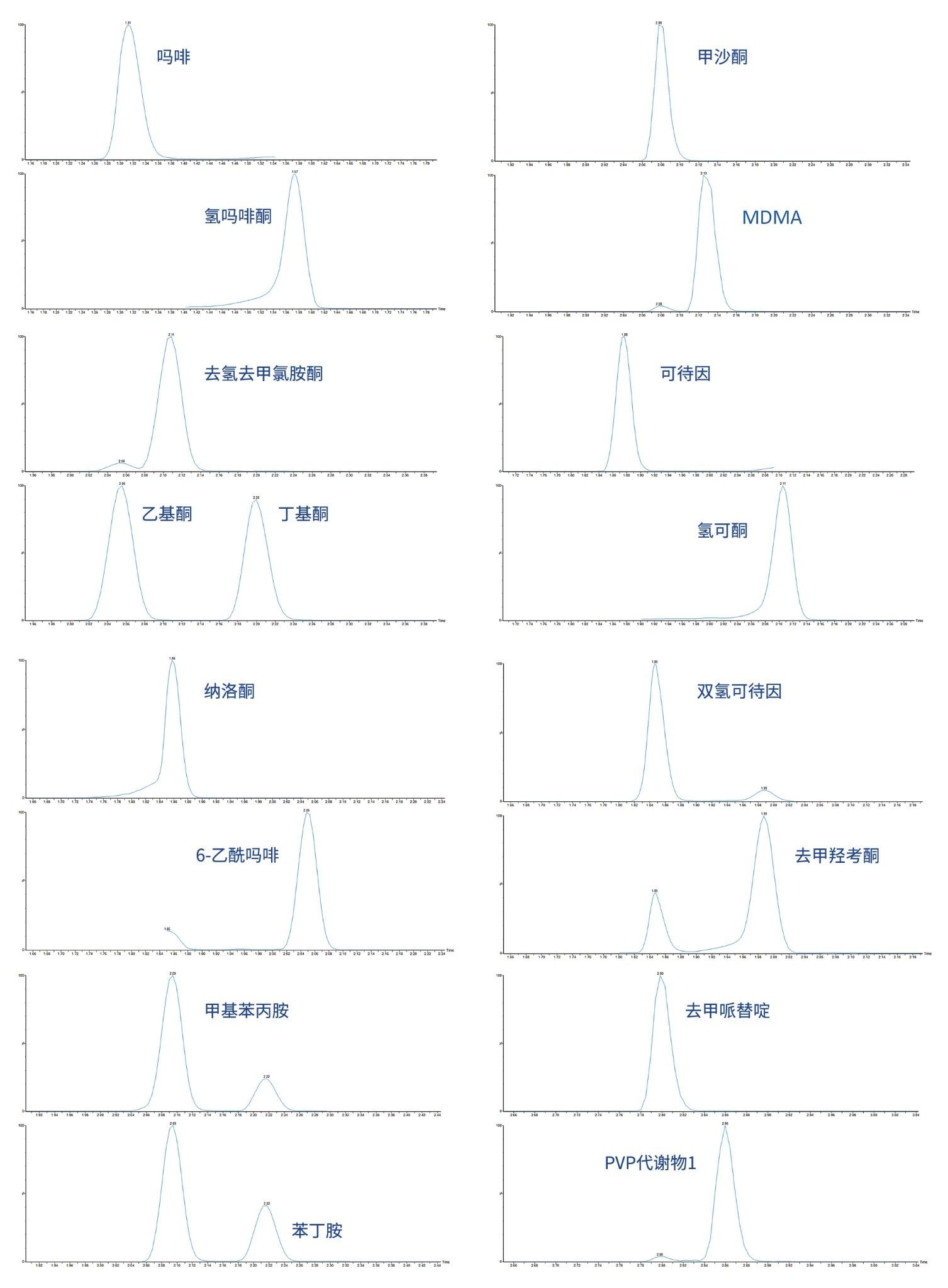
附录1中列出了所有测试目标化合物的保留时间窗口和标准曲线范围。图2显示了所有化合物的叠加色谱分离图,图3显示了几组可能相互干扰的分析物的色谱图,如下文所列。
|
吗啡和氢吗啡酮 |
甲沙酮和MDMA |
|
去氢去甲氯胺酮、乙基酮和丁基酮 |
可待因和氢可酮 |
|
纳洛酮和6-乙酰吗啡 |
双氢可待因和去甲羟考酮 |
|
甲基苯丙胺和苯丁胺 |
去甲哌替啶和PVP代谢物1 |
上述所有示例要么实现了基线色谱分离,要么采用了选择性MRM,因此这些化合物不会彼此干扰。由于本文所述的稀释法对样品净化的选择性较低,因此UPLC分离相较于应用纪要720006187ZH中略有修改。在梯度开始时设置一段临时保持时间,以便在分析物从色谱柱中洗脱之前,将盐和其他基质组分转移至废液。
每次分析均萃取两份七点标准曲线样品,较高浓度范围为25~2,500 ng/mL,中等浓度范围为10~1,000 ng/mL,较低浓度范围为2~200 ng/mL,详见附录1。所有标准曲线的相关系数(r2)均>0.99,至少75%的校正点在标称值的±15%范围内(标准曲线样品1为±20%),但N-去甲基佐匹克隆、去甲丙氧芬和佐匹克隆除外,这些化合物需要进一步研究,以确定在制备标准曲线样品时是否可以提高溶解度/稳定性。仅对N-去甲基佐匹克隆、去甲丙氧芬和佐匹克隆进行定性监测。
QC样品的较高浓度范围为25、75、187.5和1875 ng/mL,中等浓度范围为10、30、75和750 ng/mL,较低浓度范围为2、6、15和150 ng/mL,详见附录1。在运行的所有分析中,至少66%的QC样品在标称值的±15%范围内(QC1为±20%)。
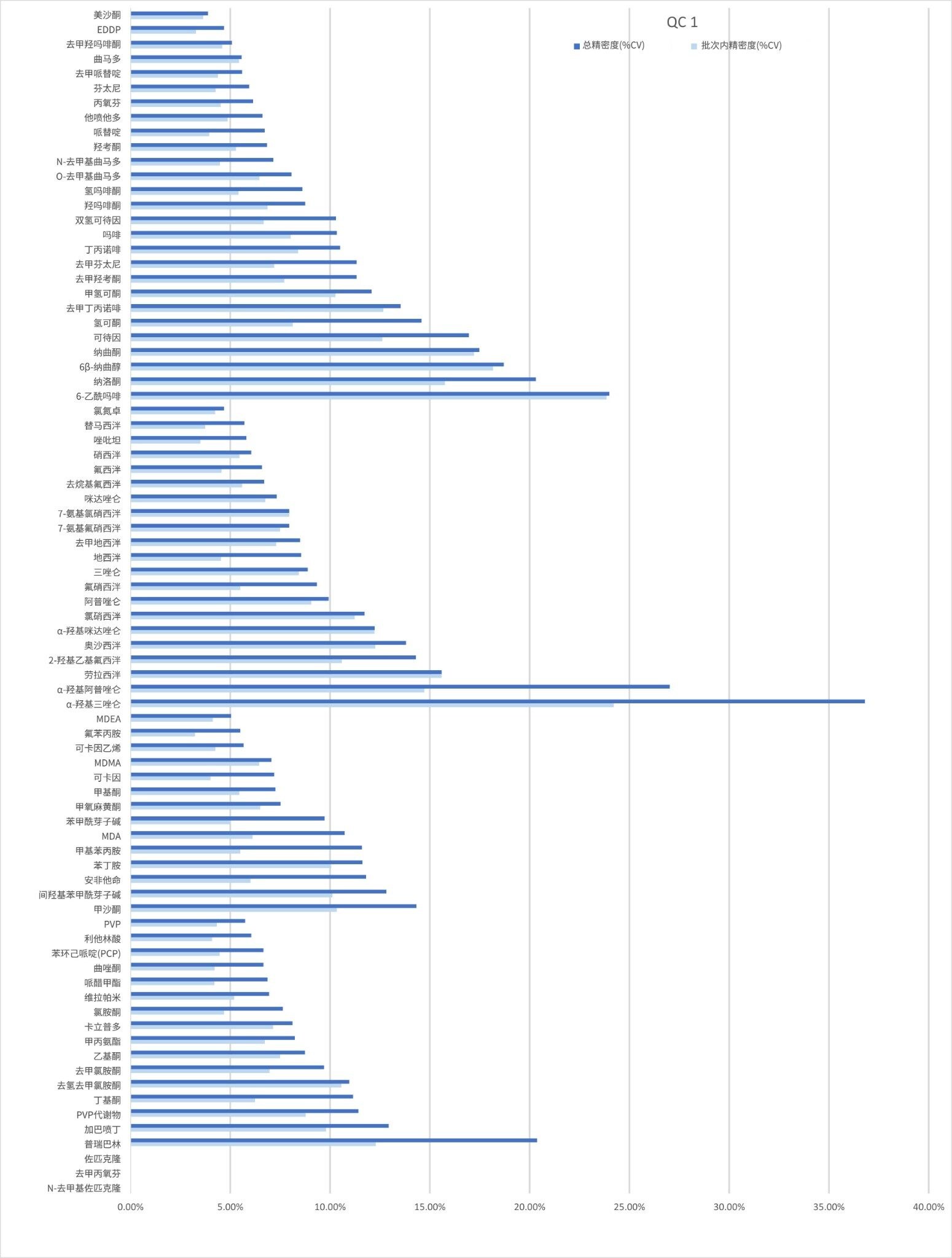
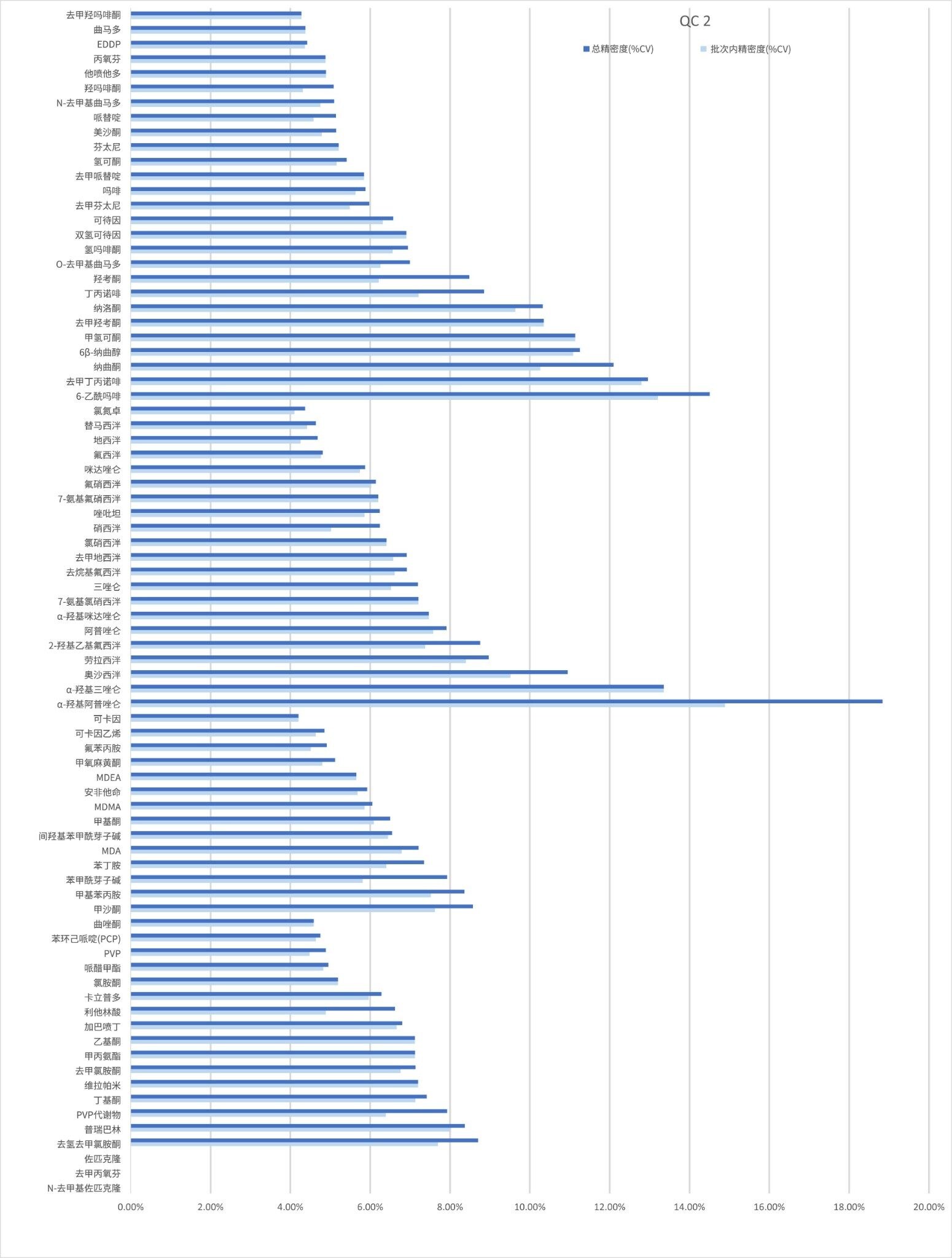
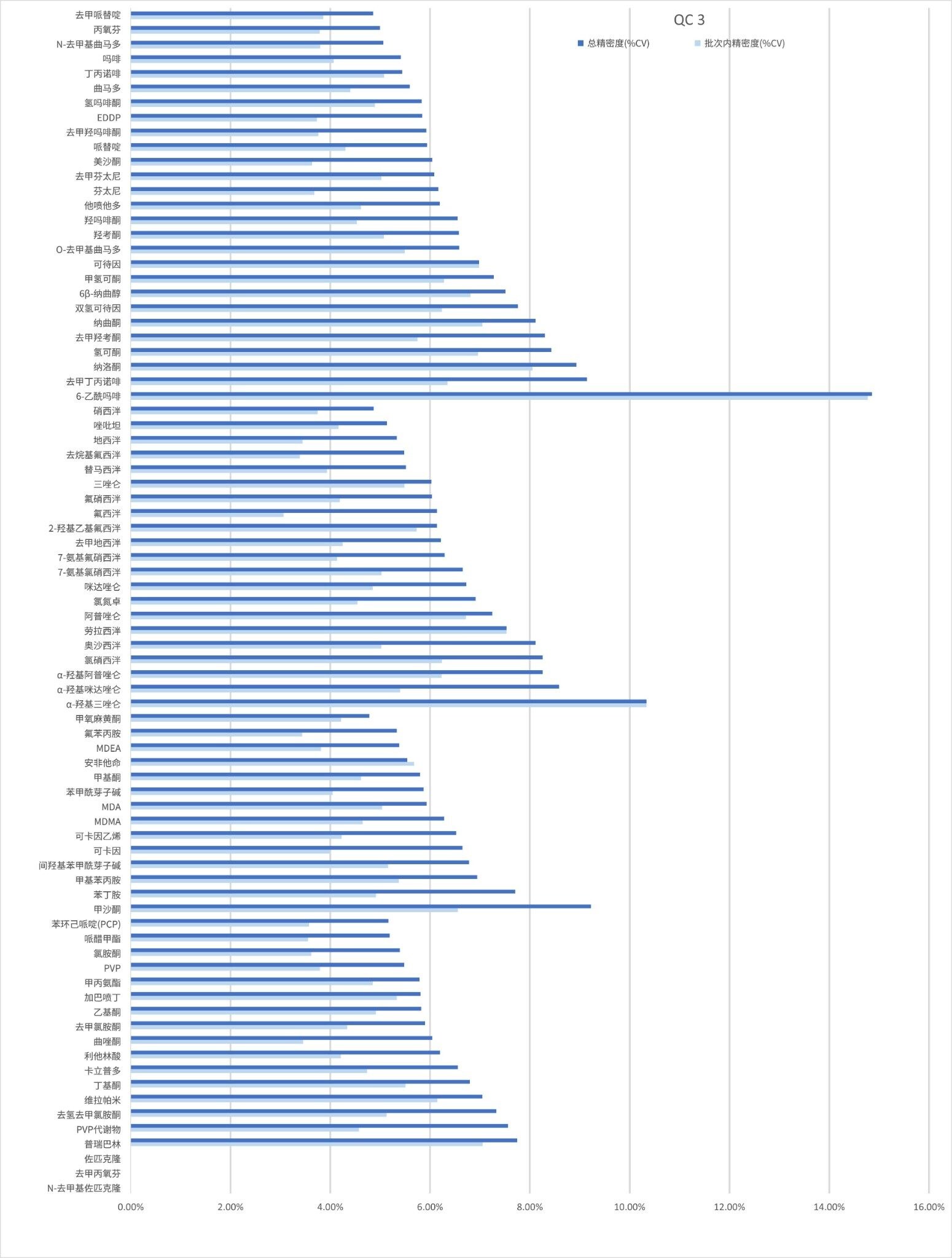
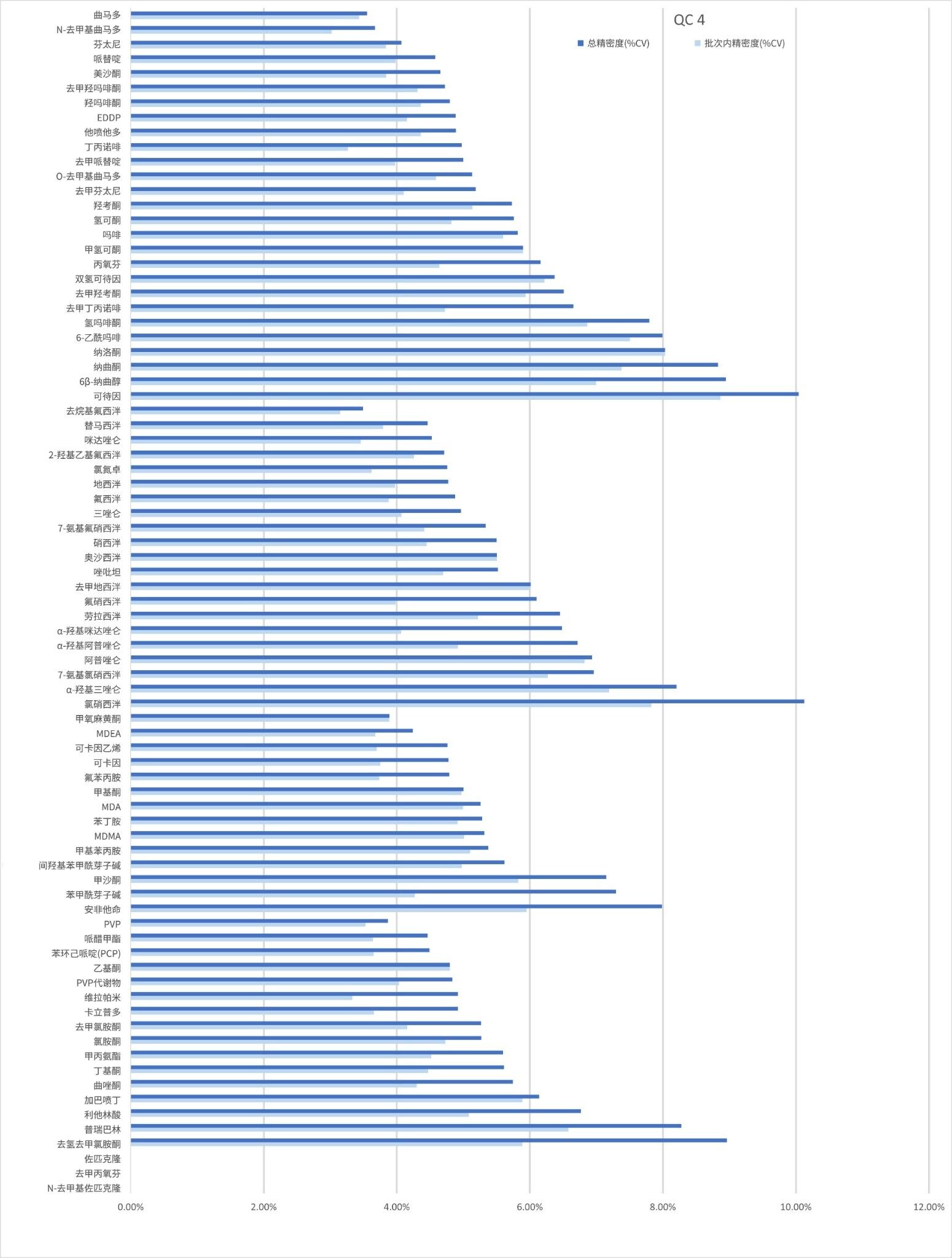
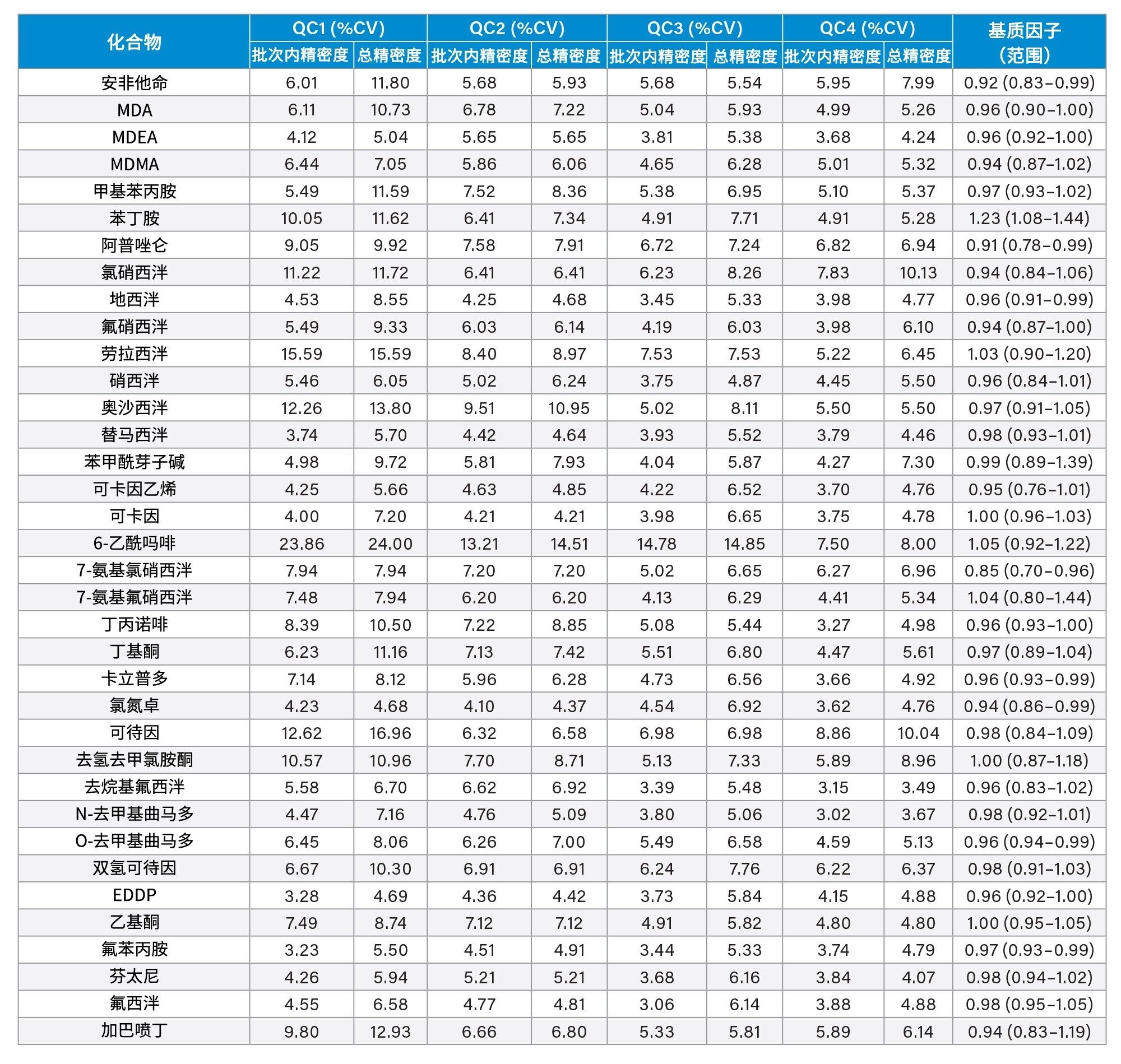
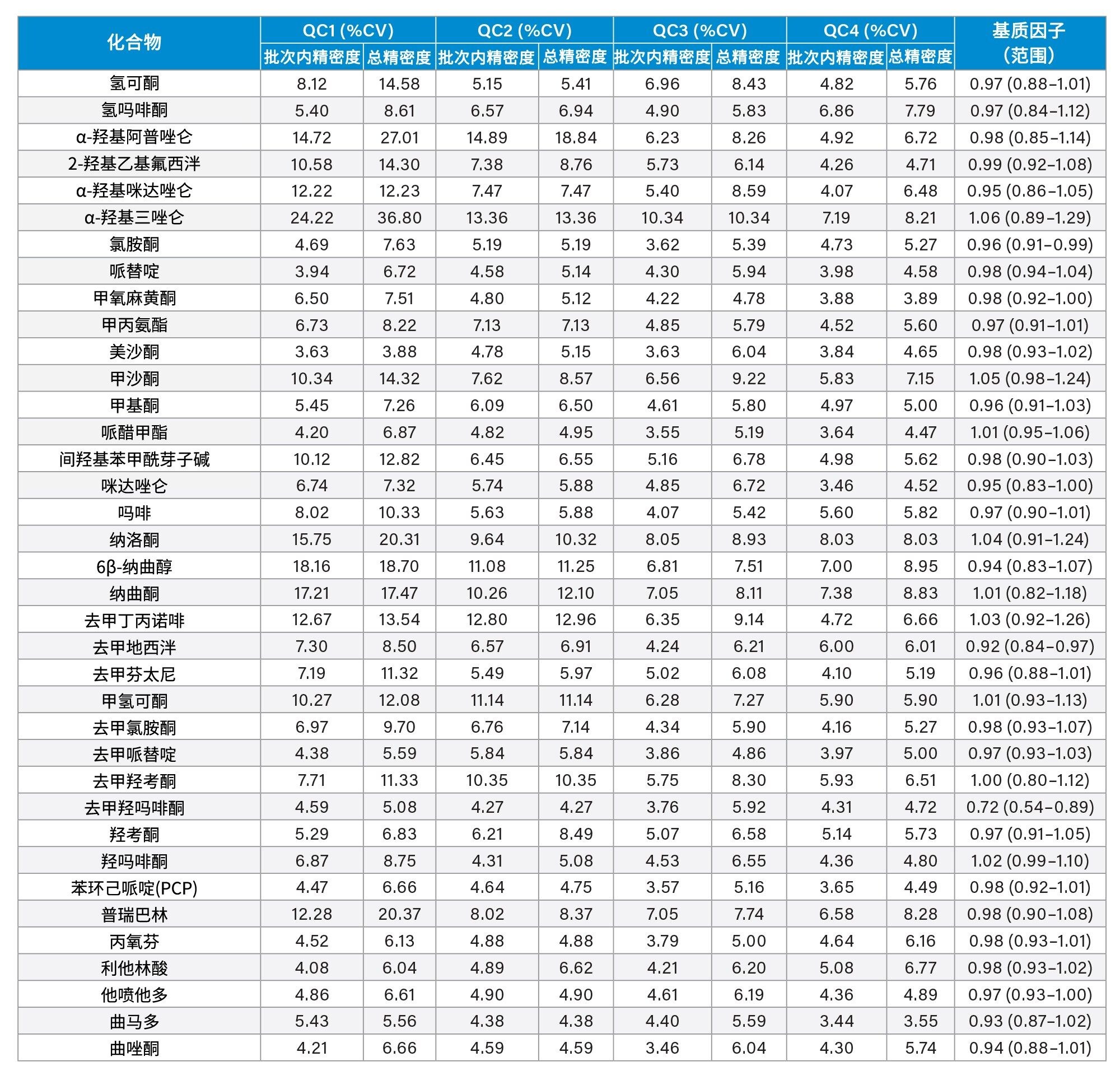
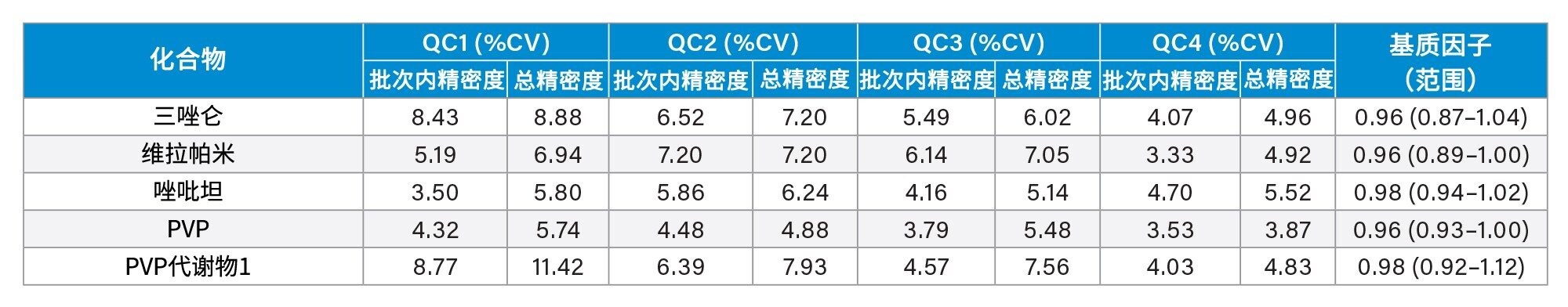
精密度性能
在五个不同的条件下,每种QC样品(浓度如上所述)重复萃取和分析五次(n=25次重复分析),评估方法精密度。图4a-4d显示了每个QC浓度下获得的结果汇总。
分析灵敏度
在四个不同的条件下,萃取并分析浓度等于或低于标准曲线样品1的低浓度样品,每种样品10个重复样,计算%CV。大多数分析物的%CV < 20%,达到可接受标准。如果劳拉西泮、6-乙酰吗啡、α-羟基阿普唑仑、α-羟基三唑仑、纳洛酮、纳曲酮和普瑞巴林不符合可接受标准,但又要求更低的浓度,则建议采用沃特世应用纪要720006187ZH中所述的样品萃取方法。
残留
在比较六个空白基质样品的平均值与浓度为标准曲线样品7两倍浓度的样品时,除了7-氨基氯硝西泮和7-氨基氟硝西泮外,其余所有分析物均未观察到显著的残留效应;这两种物质的峰面积均为标准曲线样品1峰面积的38.8%。如需评估这些分析物达到残留效应可忽略的浓度,还需要进一步测试。
基质因子
在比较从六个不同供体提取的尿液样品与水溶液对照样品,以及低浓度和高浓度加标的样品时,除了苯丙胺和去甲羟吗啡酮之外,其余所有分析物的基质因子均在0.85~1.15之间(基质效应在15%以内)。所有分析物的基质因子数据汇总见附录2。
结论
本应用简报介绍了法医毒理学研究中镇痛药物和滥用药物的UPLC-MS/MS分析概述。样品前处理流程简化为“稀释-上样”步骤,以便实施快速萃取技术。本研究所述的方法将UPLC分离与Xevo TQ-S micro IVD检测联用,能够分析大量镇痛药物和滥用药物。这个大型组中的大多数分析物都获得了高精密度的结果、一致的基质效应和极低的残留。但对于某些化合物,建议使用更彻底的样品萃取技术,以提高分析灵敏度,如沃特世应用纪要720006187ZH中所述。
参考资料
- Danaceau JP, Freeto S, Calton L. 结合简单快速混合模式的SPE与UPLC-MS/MS分析镇痛药物和滥用药物用于法医毒理学研究.沃特世应用纪要, 720006187ZH.2019年3月.
- Rosano TG, Rumberger JM, Wood M. Matrix Normalization Techniques for Definitive Urine Drug Testing.Journal of Analytical Toxicology, 2021;45:901–912.
附录1
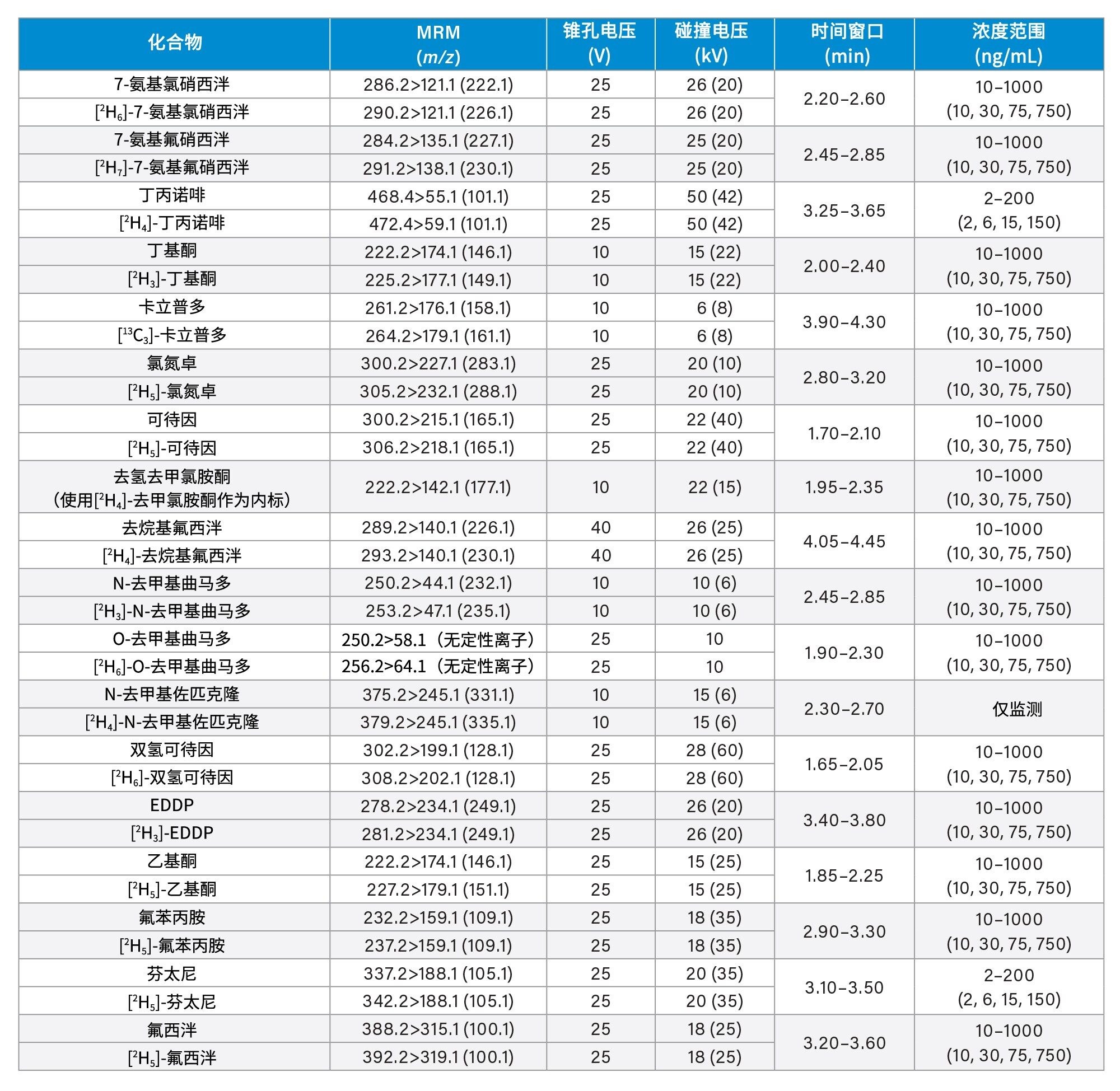
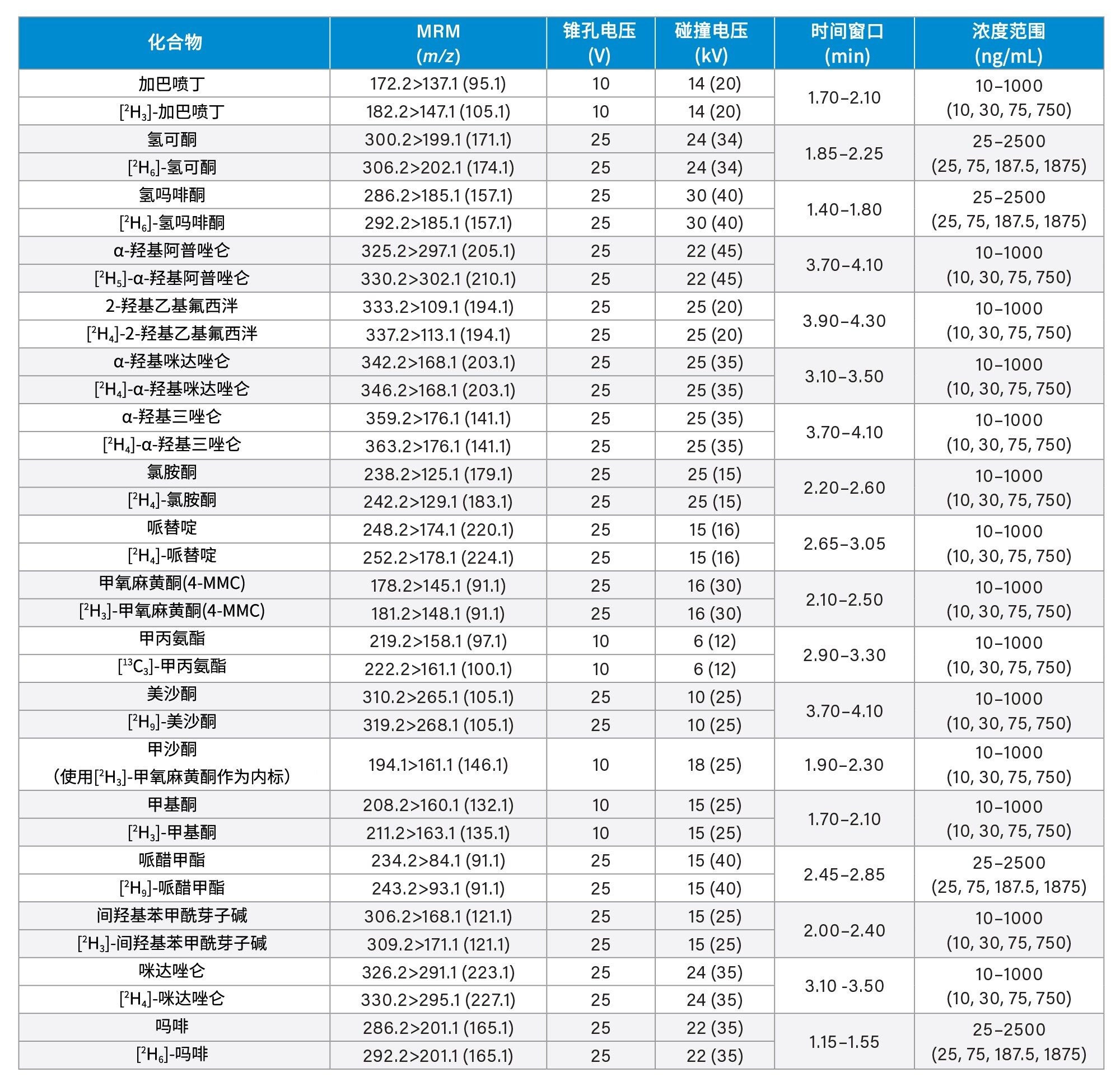
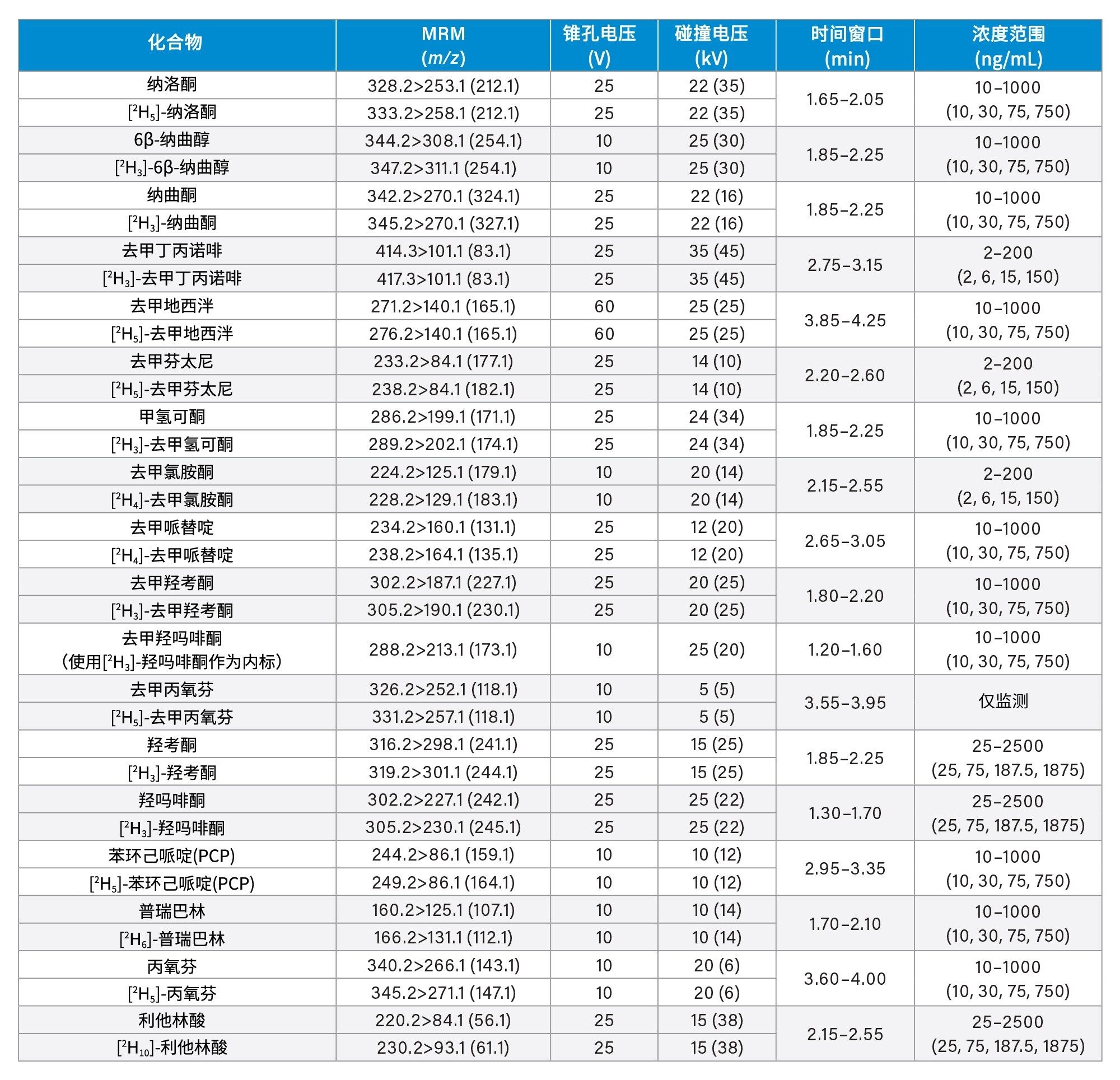
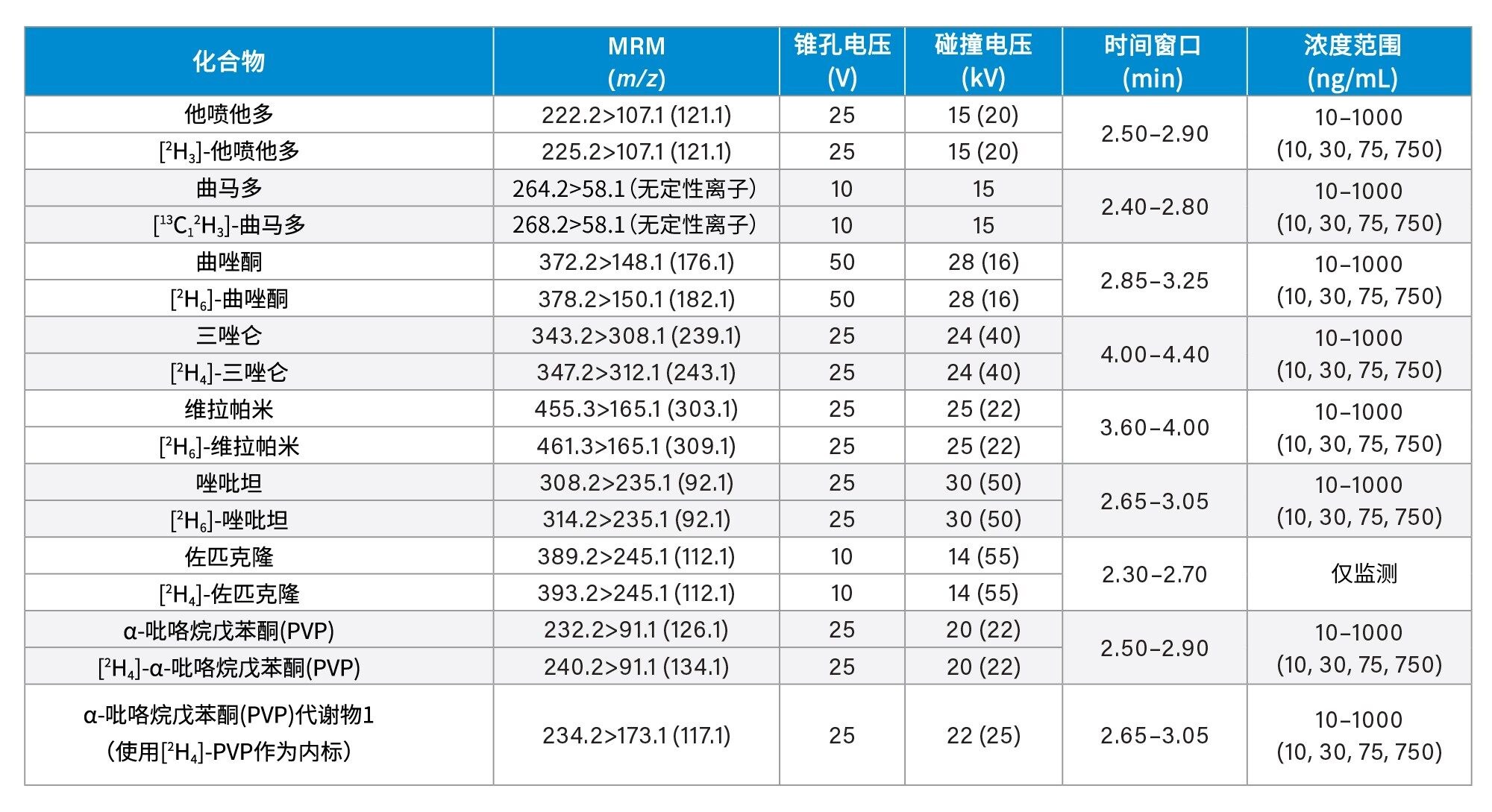
附录2
720007898ZH,2023年6月