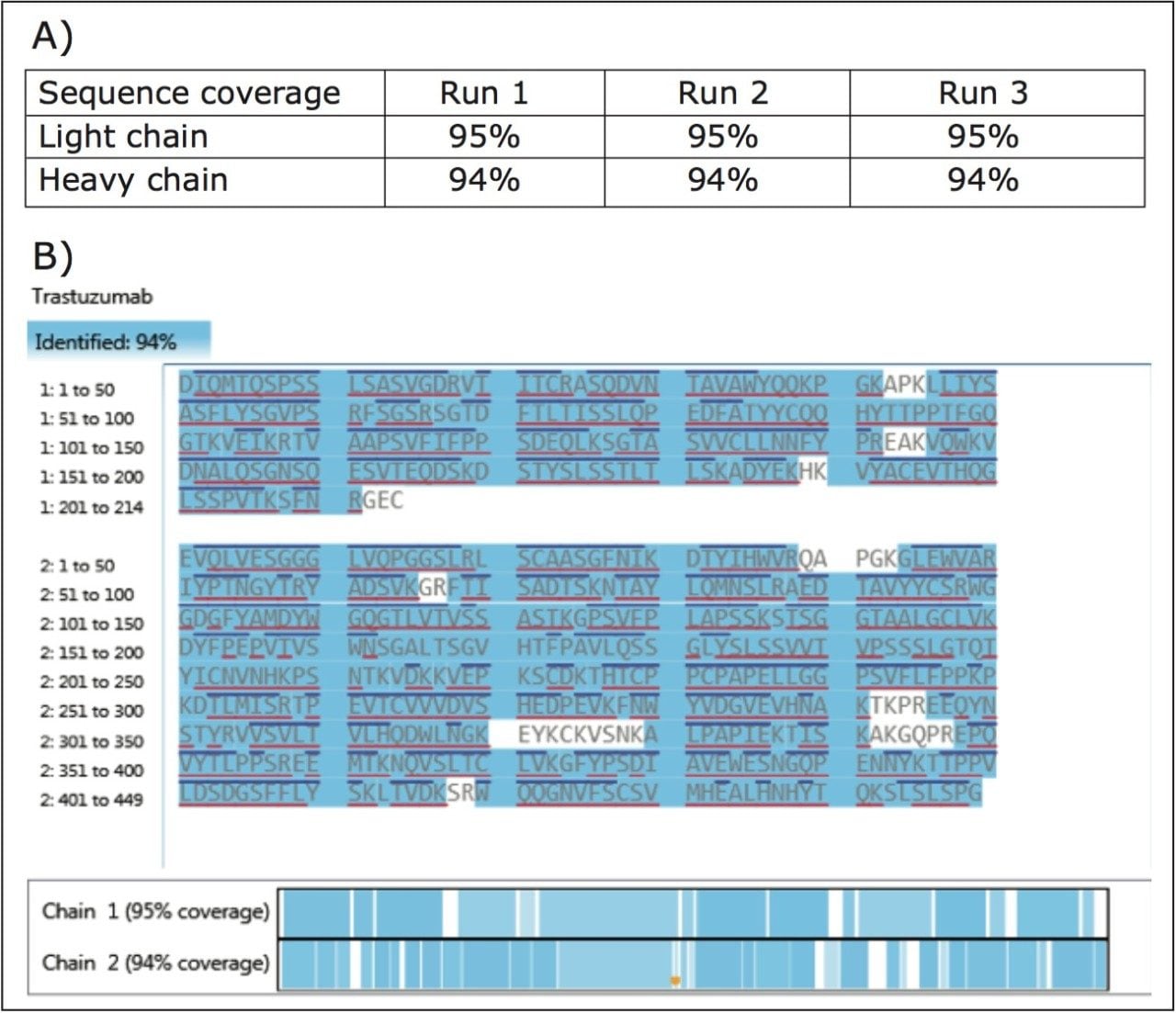
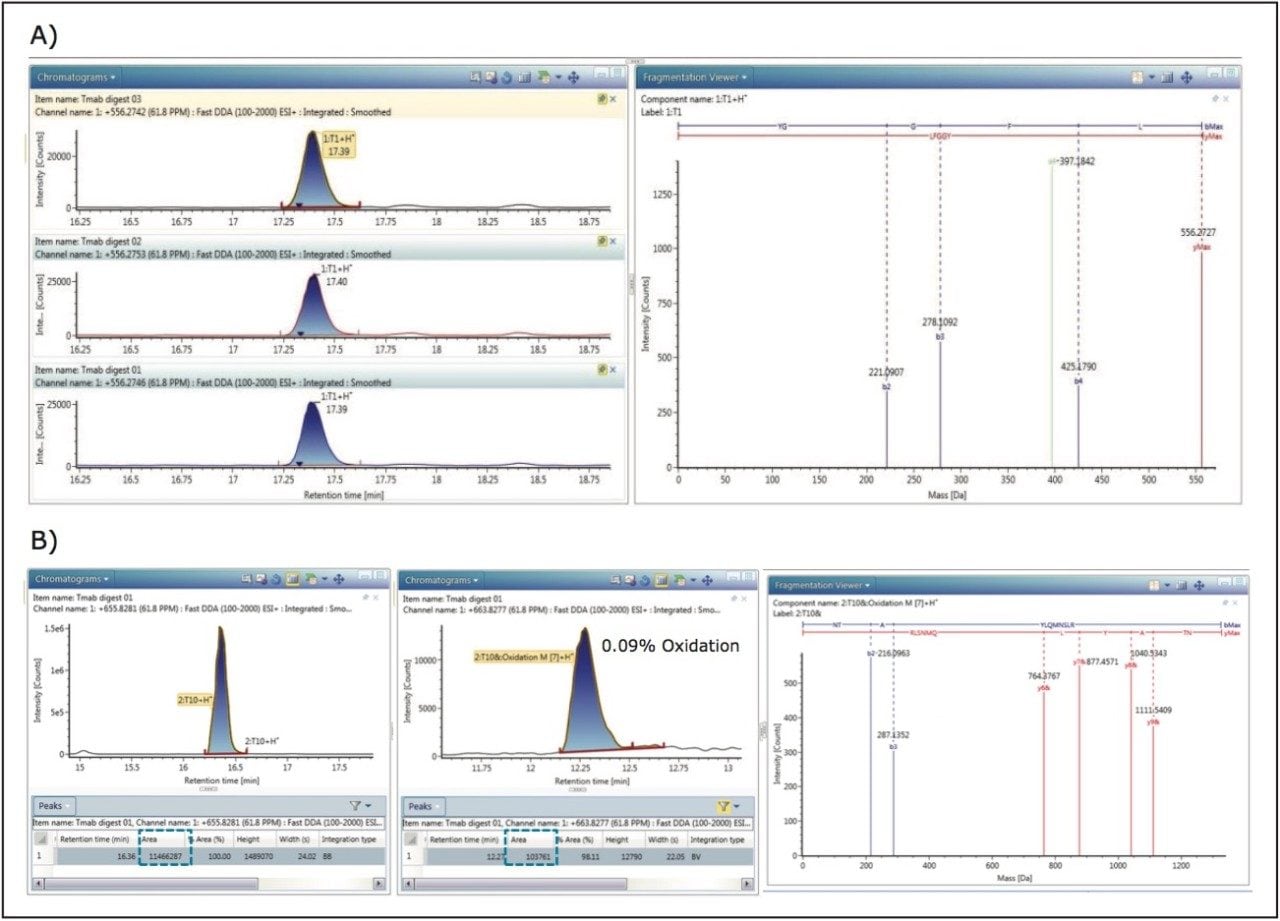
LC-MS/MS using data dependent acquisition (DDA) has been widely used to qualitatively characterize therapeutic protein digests. It is used to confirm the primary sequence of proteins and characterize their post-translational modifications (PTMs), such as oxidation, deamidation, and glycation. However, it’s also well recognized that this approach has a number of limitations, including under-sampling, a lack of reproducibility, and a lack of in-sample dynamic range. To address this, Waters offers a DDA algorithm called FastDDA. FastDDA is proven to deliver more consistent results between injections and provides greater sequence coverage of proteins.1
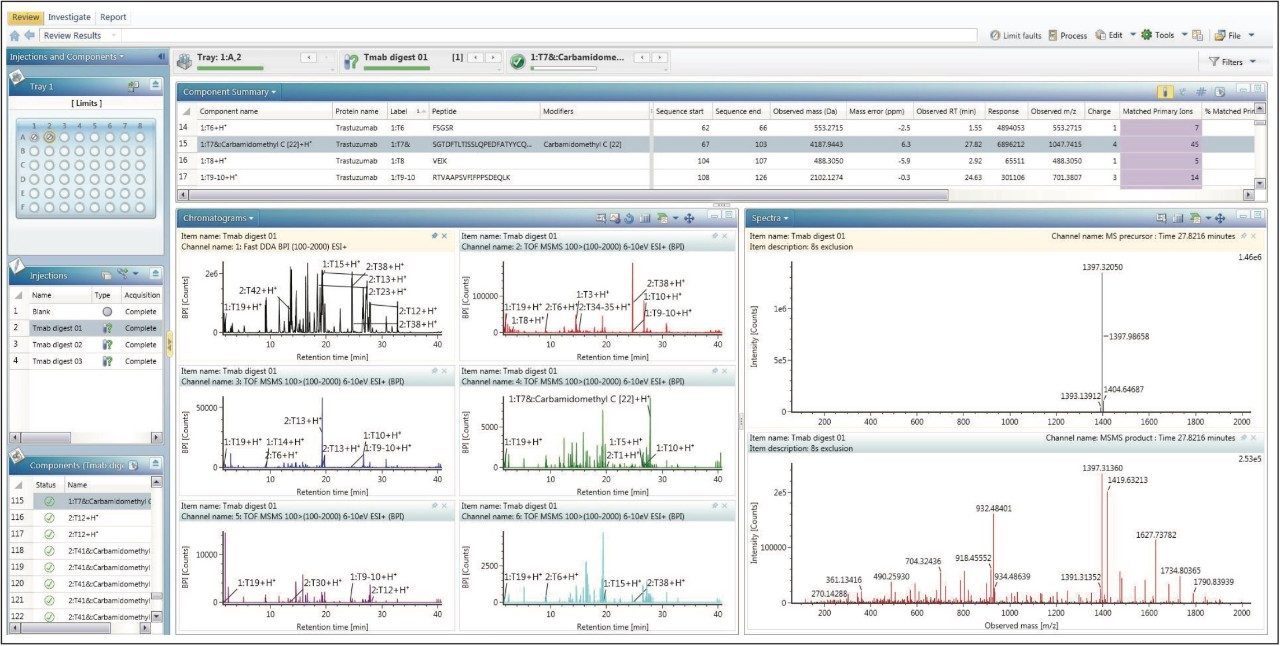
The Biopharmaceutical Platform Solution with UNIFI provides a dedicated DDA peptide mapping workflow, which streamlines data acquisition, processing, and report generation. This streamlined workflow enables an efficient way to analyze a large batch of samples and compile complex results into a comprehensive report.
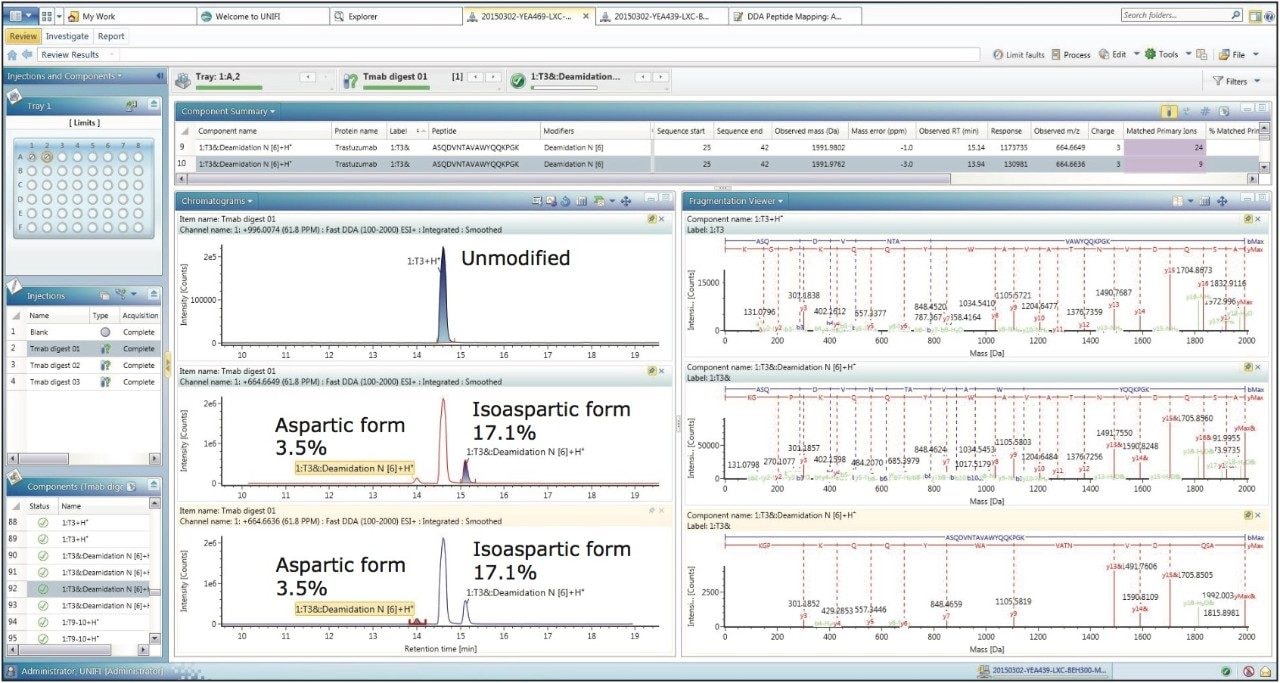
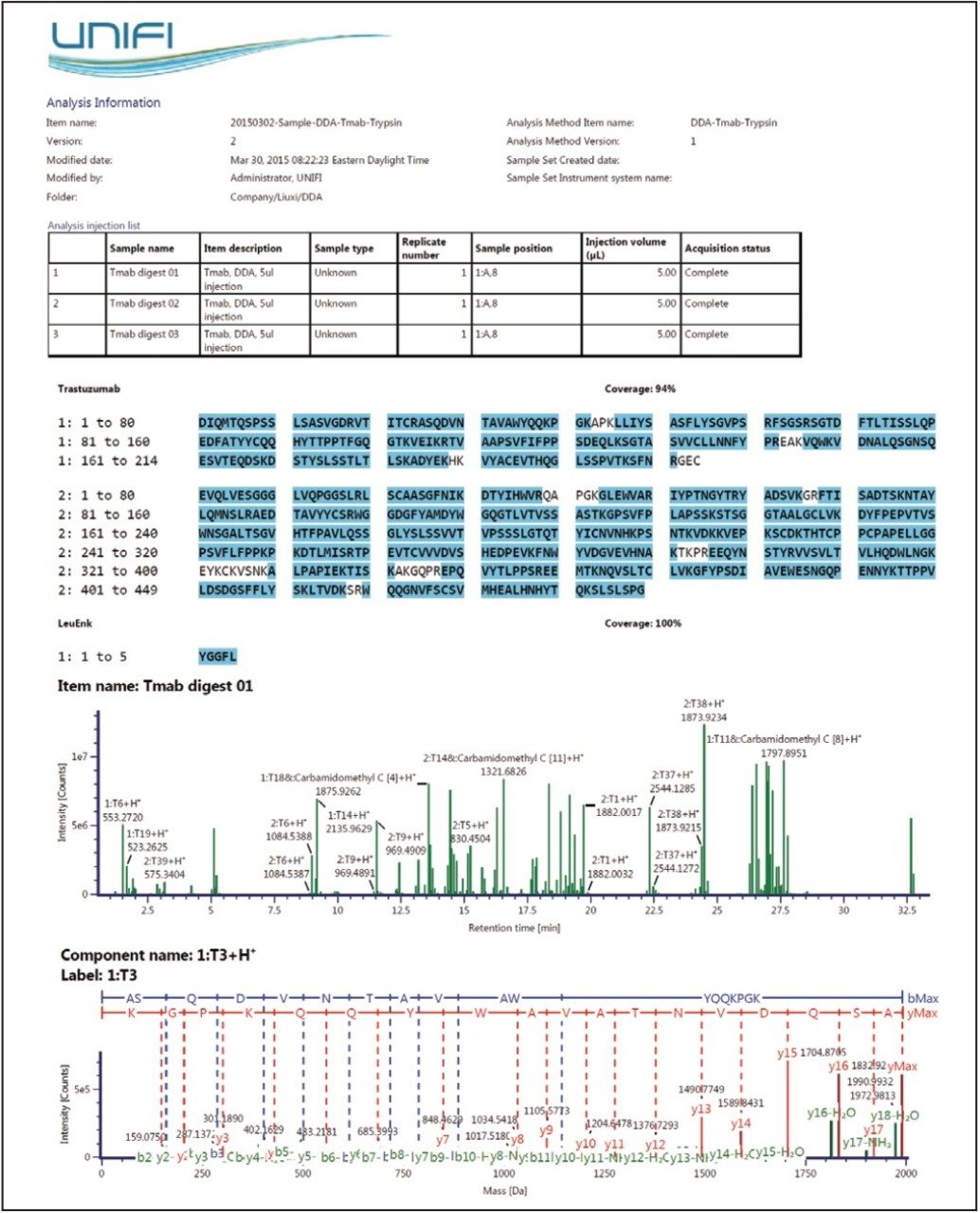
Here, we illustrate this workflow using a trastuzumab tryptic digest mixture, which has been spiked with a standard peptide leucine enkephalin at 0.1% level. The platform used for this study is comprised of an ACQUITY UPLC H-Class Bio System and an ACQUITY UPLC Tunable UV (TUV) Detector in-line with a Xevo G2-XS QTof Mass Spectrometer. The Xevo G2-XS QTof combines StepWave ion optics with an XS collision cell, and significantly increases MS and MS/MS sensitivity without reduction in selectivity.2,3
In this study, we evaluate the MS and MS/MS data quality, especially for low abundant peptides, and the reproducibility of the FastDDA method. In addition, we demonstrate the informatics tools within the UNIFI Scientific Information System that enable efficient reviewing and reporting of the DDA peptide mapping data.