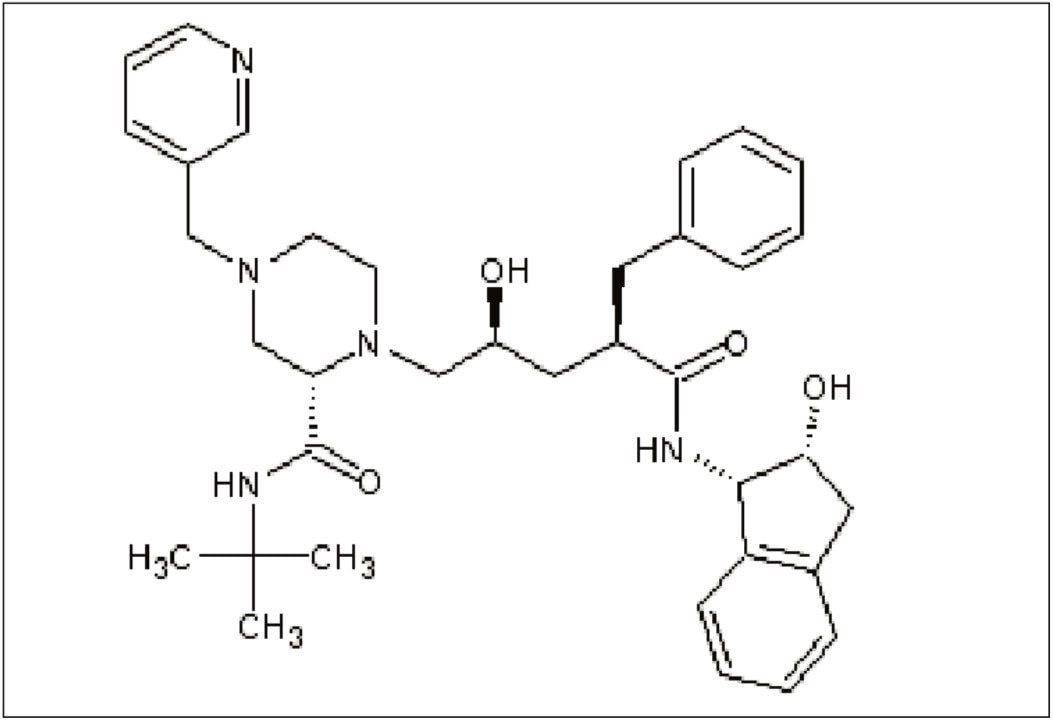
Metabolite identification (Met ID) plays a crucial rule throughout the drug discovery and development process. The key analytical requirements for an effective Met ID study are: high chromatographic resolution with a fast turnaround time, metabolite identification with positive confirmation, and comprehensive structure elucidation.
Waters UltraPerformance LC (UPLC) technology has been gaining wide acceptance throughout the pharmaceutical industry since its first commercial instrument, the ACQUITY UPLC System, was introduced in 2004. The system’s ability to operate at 15,000 psi (1000 bar) backpressure enables the use of the sub-2 μm particle columns. The optimal linear velocity of this particle size extends far higher than that of the conventional 3 μm or 5 μm column packing. This higher linear velocity produces chromatographic separation with significantly enhanced resolution, speed, and sensitivity.
Waters SYNAPT MS TOF-based mass spectrometers are rapidly becoming the preferred analytical tool for Met ID studies. Its high full-scan sensitivity and exact mass measurement capability greatly enhance metabolite identification. In addition, the rich fragmentation information provided makes the task of localizing the site of biotransformation easier and more precise.
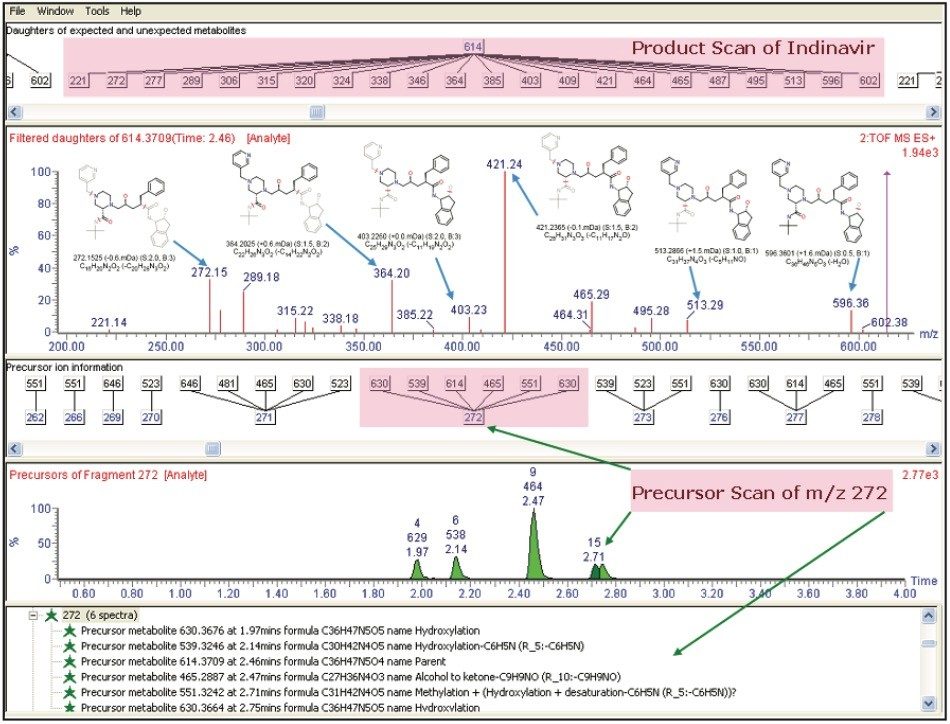
Finding metabolite “soft spots” is often a major bottleneck for the Met ID process. It is beneficial to have a tool that enables the scientist to take advantage of the information-rich data provided by the UPLC/Synapt MS system to rapidly find these soft spots.
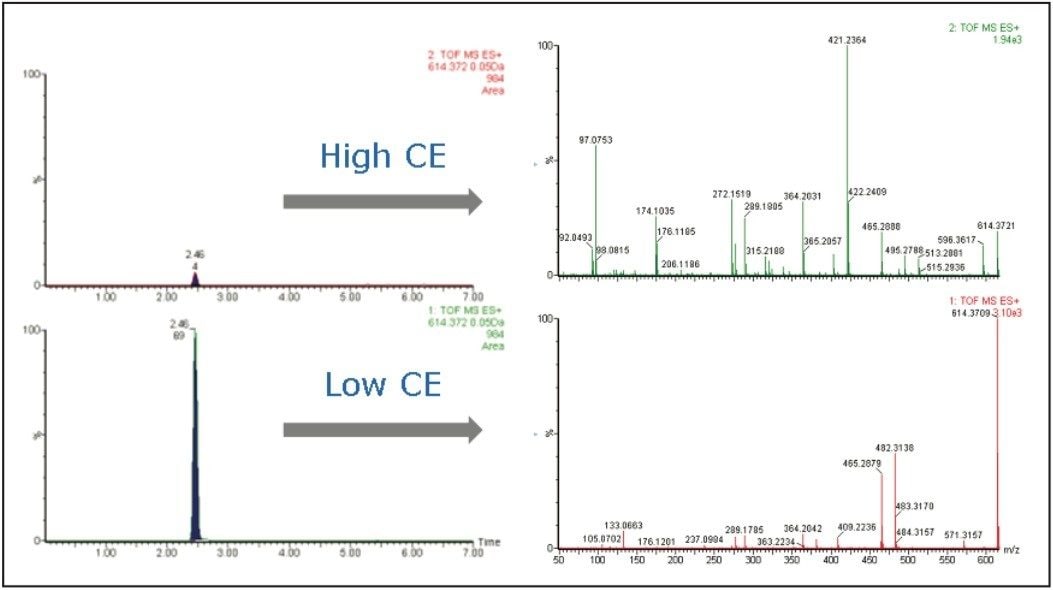
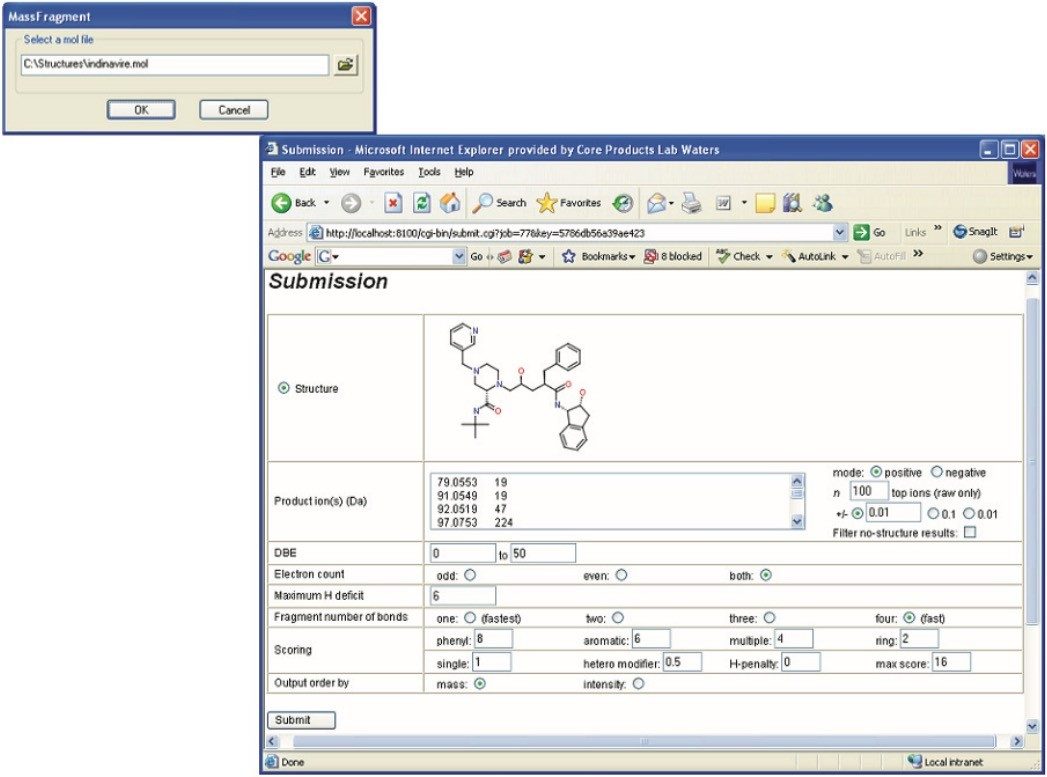
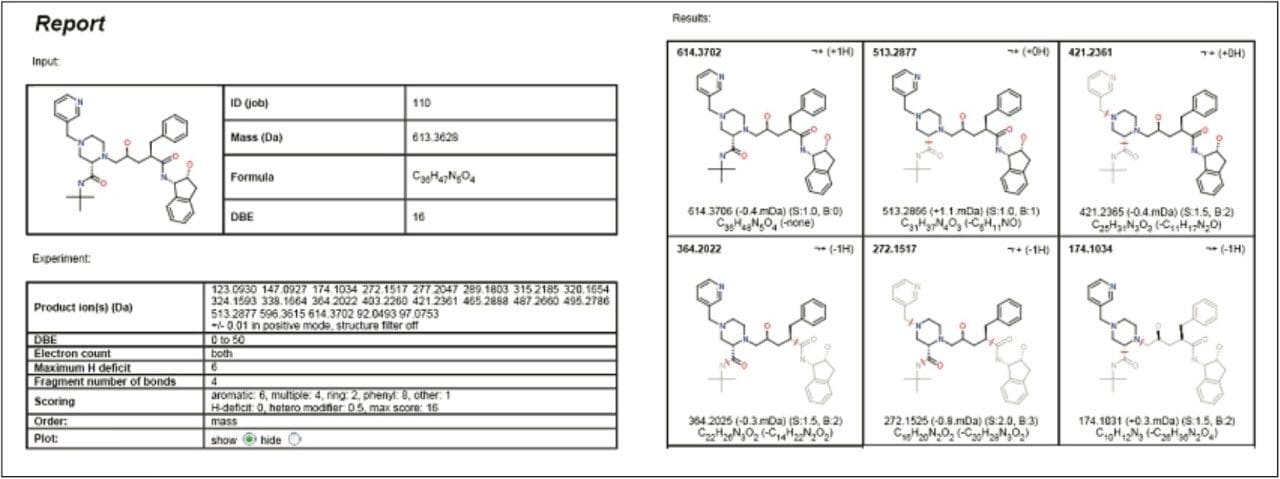
This application note demonstrates the use of a UPLC-SYNAPT MS system employing the MSE strategy for metabolite identification. Using the TOF MSE, the LC-MS data from the low collision energy (CE) MS scan and high CE MS scan can be both obtained during a single UPLC injection1-2. The structural elucidation was easily performed with the aid of a software tool, MassFragment, by directly importing the fragment ions from raw data file or from the MetaboLynx Application Manager’s MSE Fragment Analysis window.