The increasing complexity of bottom-up proteomics challenges the capabilities of mass spectrometers to generate more and more detailed information. Instrument speed, sensitivity, and mass accuracy have all increased significantly over recent years, thereby affording better quality data, improved peptide sequence annotation, and more accurate identification results.
In line with improved hardware features, novel LC-MS acquisition schemas and fragmentation mechanisms have been introduced, including parent ion discovery (PID) methods, data independent acquisitions (DIA), ion mobility (IM) assisted methods, and electron transfer dissociation (ETD). To date, IM has been mainly employed for the cross sectional and structural analysis of various analyte types,1 and enhancing the specificity of DIA acquisitions such as HDMSE.2
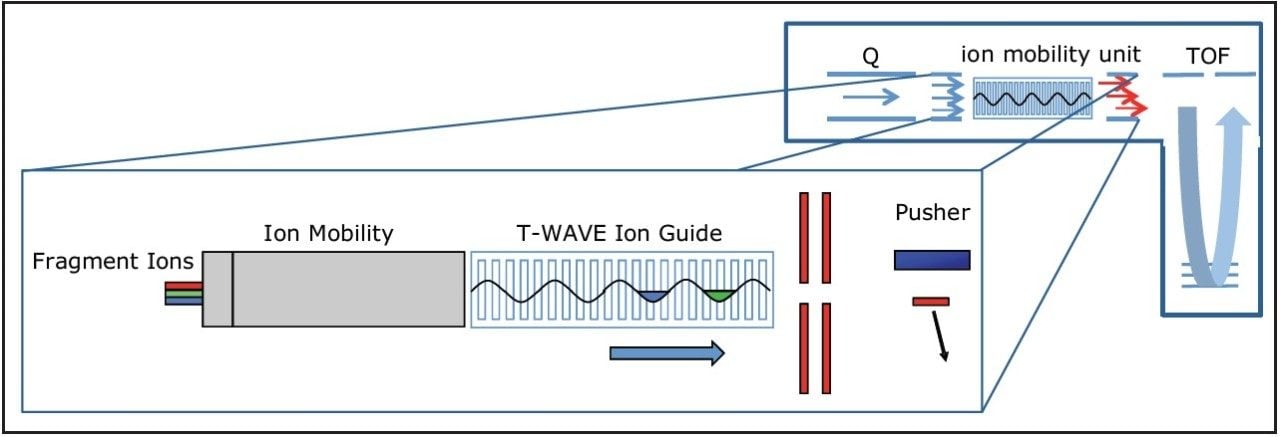
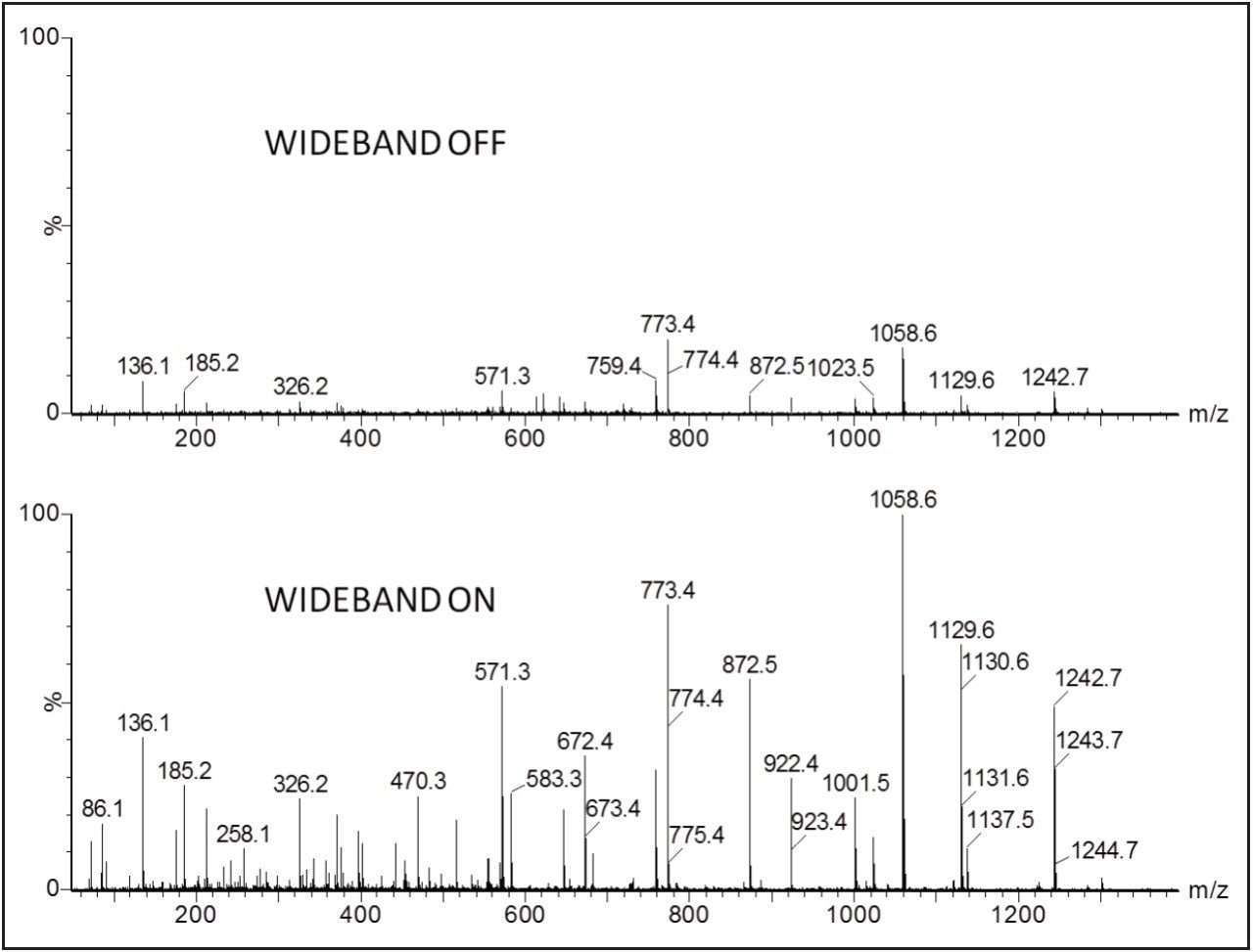
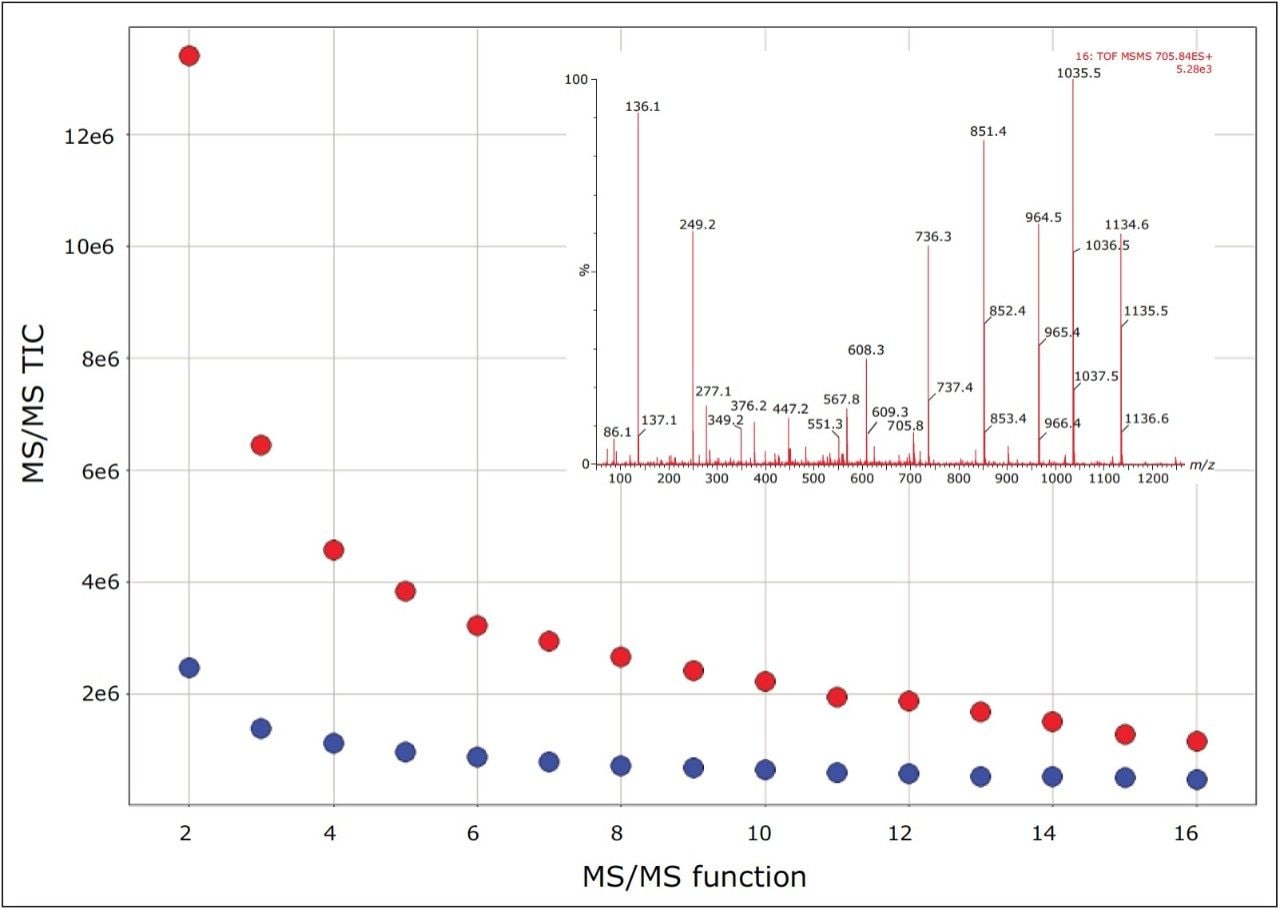
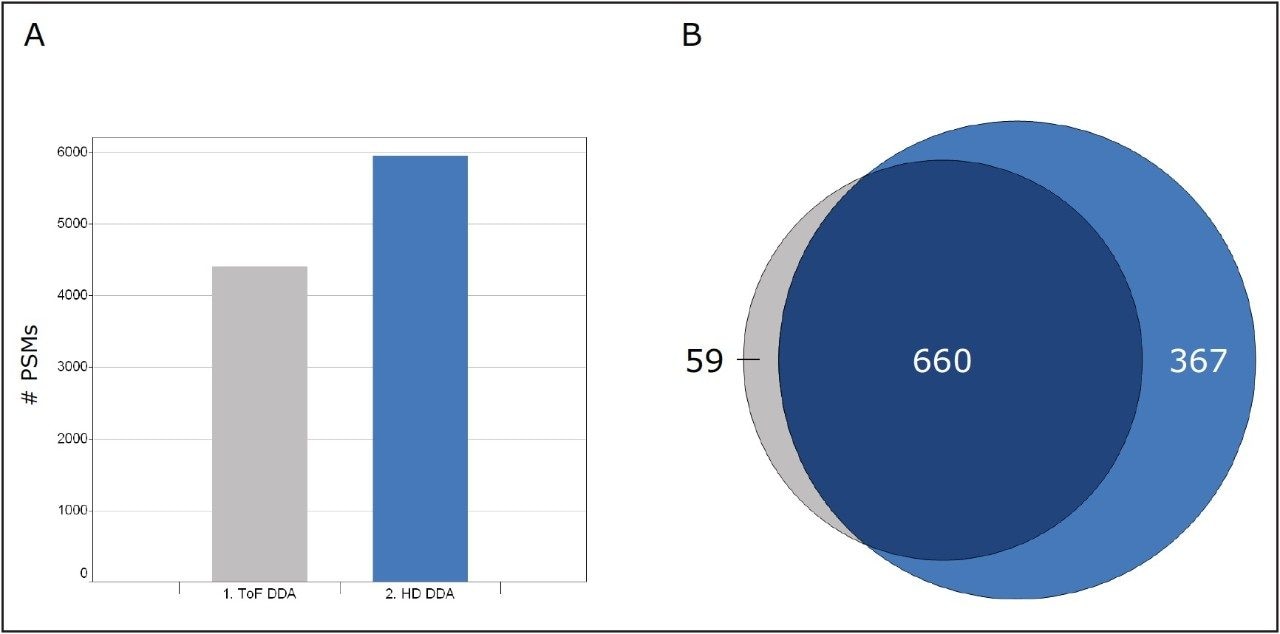
In this study, instrumental and application benefits are demonstrated for the identification of proteins and peptides in a new high definition data directed analysis (HD-DDA) mode, where ion mobility spectrometry is incorporated into a quadrupole time-of-flight mass spectrometer. HD-DDA uses a high duty cycle mode and enhanced decision making to provide a highly sensitive and selective experiment.