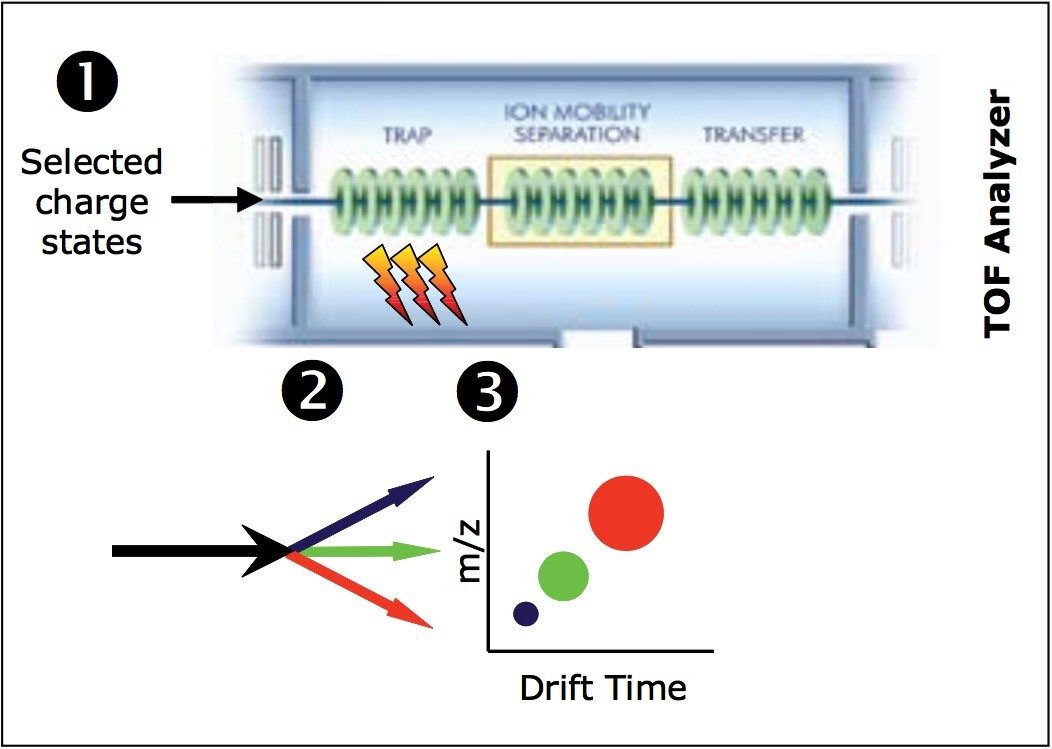
The Waters SYNAPT High Definition Mass Spectrometry (HDMS) System combines high-efficiency ion mobility with high-performance tandem mass spectrometry. This enables the analysis of samples differentiated by size, shape, and charge, as well as mass, to deliver increased specificity and sample definition.
In this study, we describe the use of the SYNAPT HDMS System to quickly obtain a partial amino acid sequence of a monoclonal antibody (mAb) via a top-down approach.
In this method, reduced mAb is introduced to the SYNAPT HDMS System via on-line desalting. Selected charge states of the intact light chain subunit are subjected to collision-induced dissociation (CID) prior to separation of the resulting fragment ions based on their gas phase mobilities.
Post-acquisition data processing produces simplified top-down fragmentation spectra containing fragment ions predominantly from one charge state, from which N-terminal amino acid sequence (up to 11 residues) of IgG can be readily deduced.
This structural information, coupled with molecule weight measurement of the intact mAb, can be used to support comparability studies and as a rapid ID test, reducing testing times and allowing for more efficient manufacturing processes.