A Simple Dilute and Shoot Method for the UPLC-MS/MS analysis of Pain Management Drugs and Drugs of Abuse From Urine for Forensic Toxicology
Ce document est une note d’application et ne contient pas de section détaillée concernant l’expérimentation.
For forensic toxicology use only.
Abstract
This application brief describes a simple dilute and shoot method for the UPLC-MS/MS analysis of pain management drugs and drugs of abuse for forensic toxicology and offers an alternative approach to that described in Waters Application Note. 720006187. Sample preparation is simplified from SPE to a single dilution step, which still provides minimal carryover, consistent matrix effects, and precise quantitative data, reaching the required analytical sensitivity for the majority of analytes within the panel. For the compounds requiring more analytical sensitivity, further sample clean up, as described in the original application note, is recommended.
The use of an ACQUITY™ UPLC™ BEH™ C18 Column allows for a fast analysis of a large panel of compounds, while maintaining all required separations to ensure no interference from isobaric compounds.
The Waters™ Xevo™ TQ-S micro IVD provides accurate quantification of the large panel of analytes over wide dynamic ranges, simultaneously quantifying some analytes at 2 ng/mL with others at 2500 ng/mL.

The combination of the sample extraction, chromatography separation and MS/MS detection gives a simple workflow, giving a fast and precise method that can also be automated on a Hamilton STAR or STARlet system.
Benefits
- Fast and simple method for the analysis of a comprehensive panel of definitive drugs
- Simple dilute and shoot sample extraction method
- Consistent matrix effects and recoveries with minimal carryover
- Precise quantification of a large panel of definitive drugs
Introduction
Analyte panels for use in forensic toxicology analysis typically include illicit drugs and common drugs of abuse. Often, multiple methods are used to obtain a comprehensive view of the multiple drug classes. These methods may include immunoassay, GC-MS, LC-MS/MS, or a combination of methods. Waters has developed a method for the quantification of a comprehensive drug panel to achieve the appropriate analytical sensitivity, selectivity, and accuracy for unambiguous identification for forensic toxicology.
This method employs a simple sample extraction procedure using a dilute and shoot approach coupled with a rapid and reproducible chromatographic method using an ACQUITY UPLC BEH C18 Column that achieves baseline separation for all critical pairs of potentially interfering analytes. A Waters Xevo TQ-S micro IVD Mass Spectrometer with Xtended Dynamic Range (XDR) capabilities provided the analytical sensitivity and dynamic range capabilities required for this diverse group of compounds. Although this method has been shown to provide suitable results for pain management drugs and drugs of abuse, there are some limitations as a result of the simplified dilute and shoot workflow around analytical sensitivity. If low analytical sensitivity is required for these small number of analytes, it is recommended that further sample clean-up is required such as that described in Waters Application Note. 720006187.
Experimental
Sample Extraction
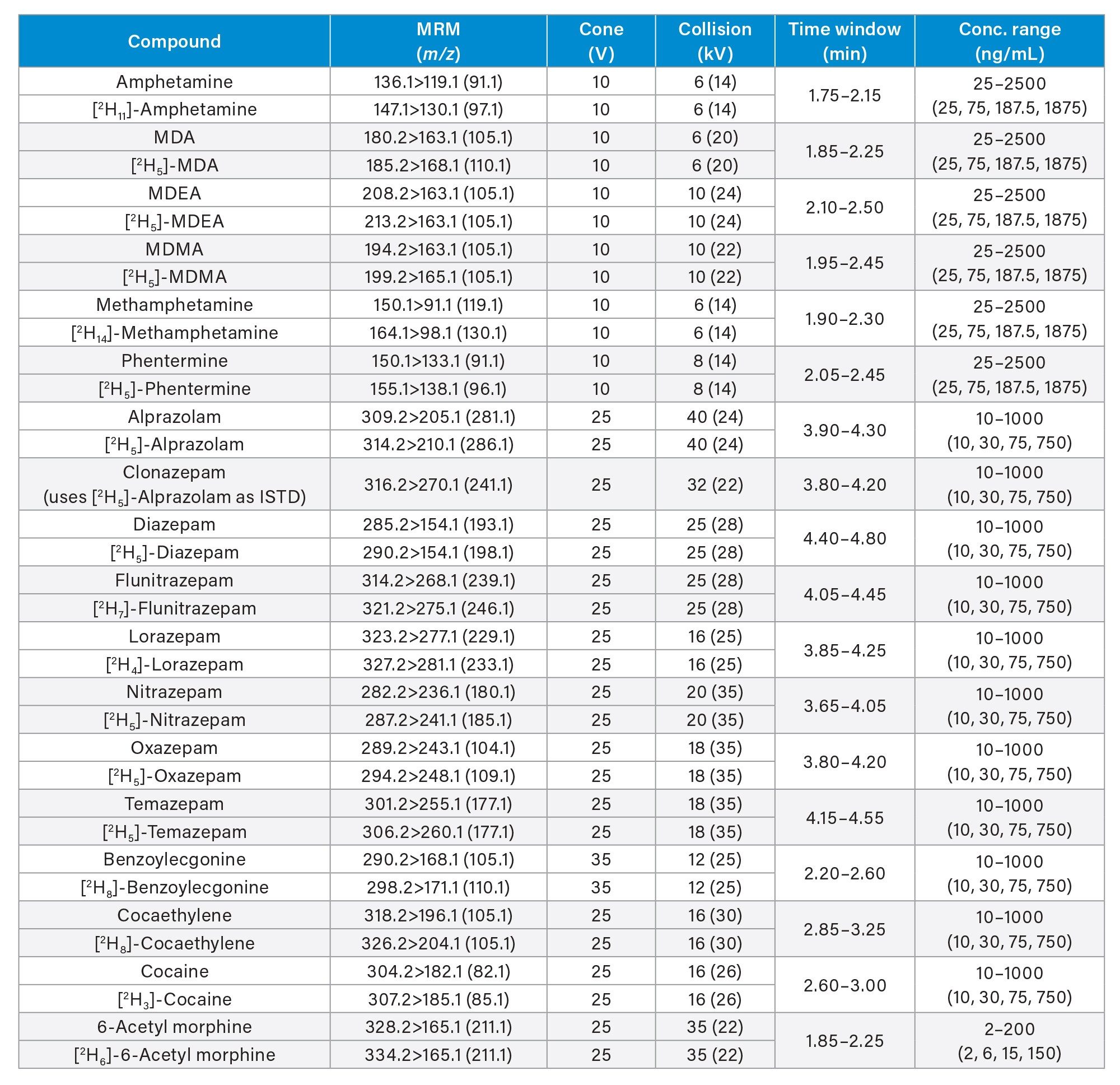
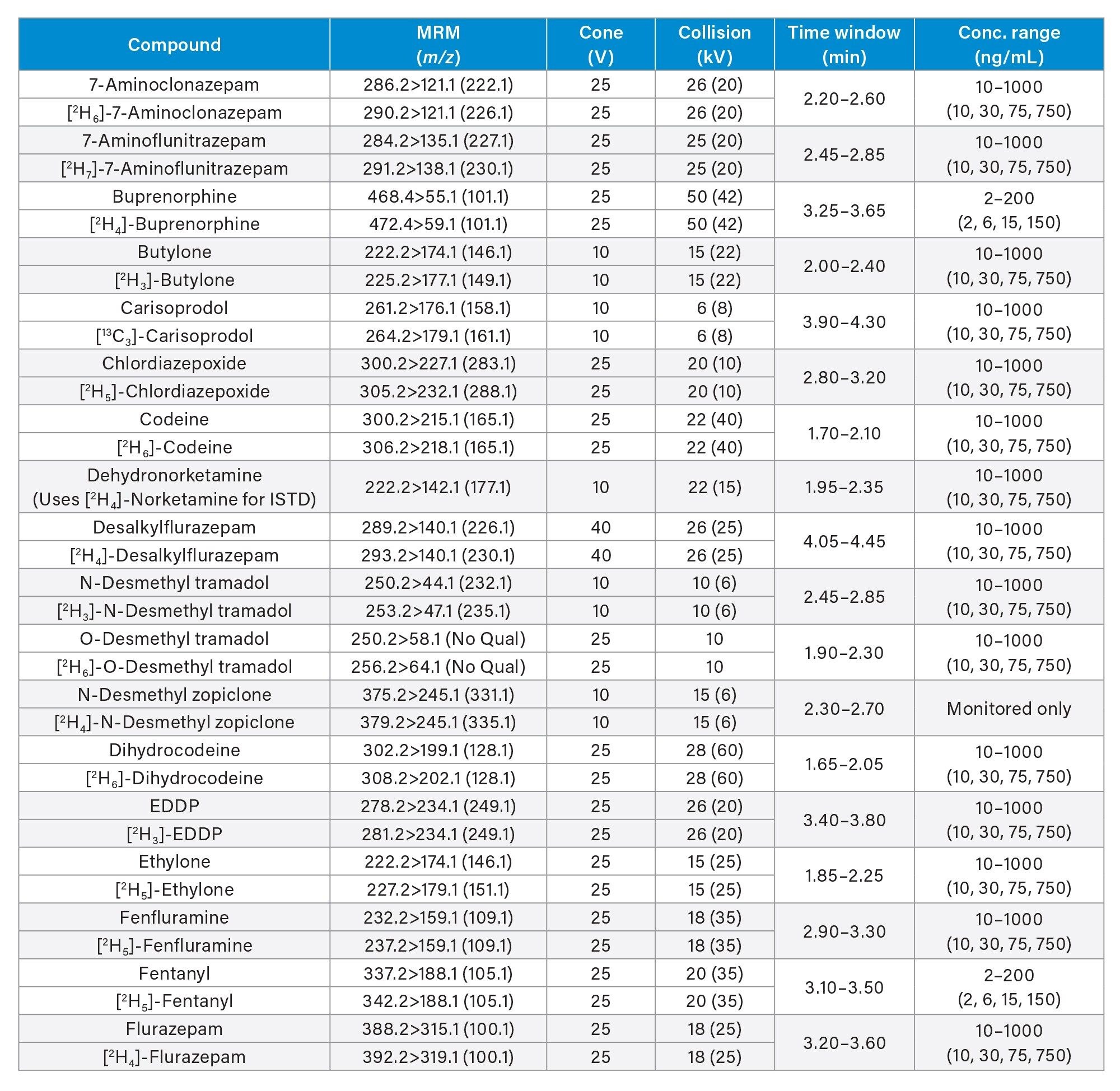
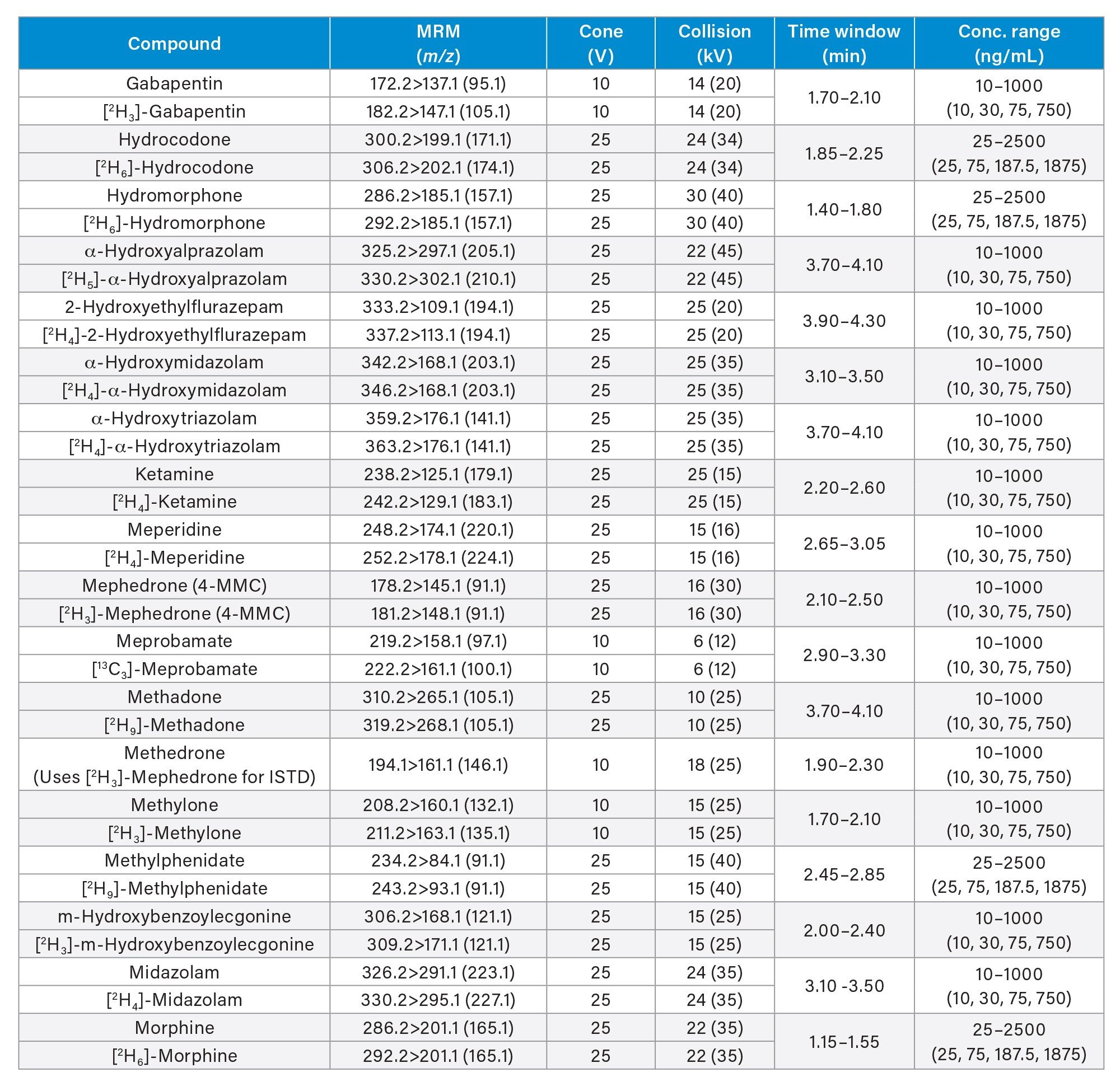
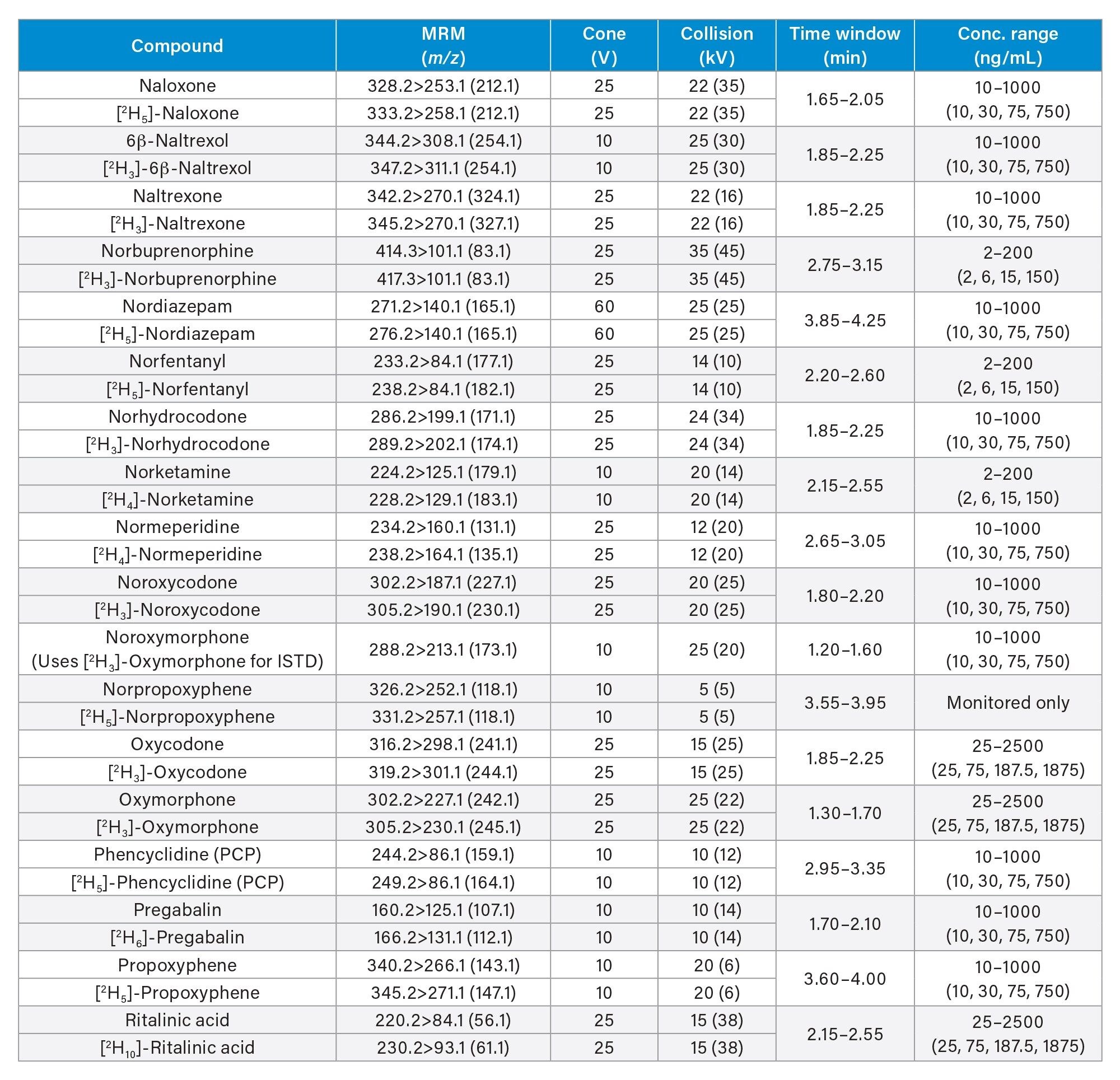
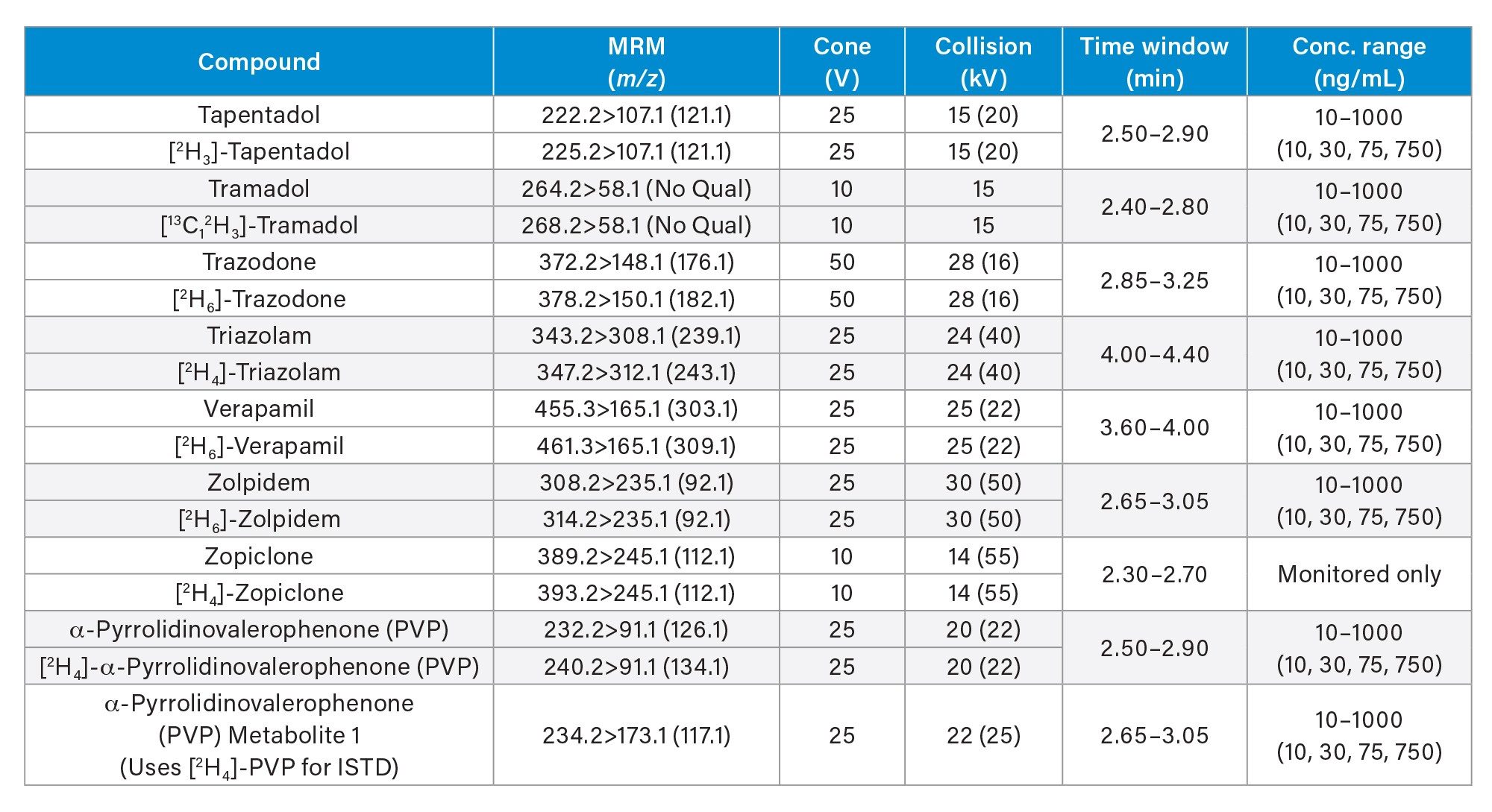
All standards were obtained from Cerilliant (Merck Life Sciences, Gillingham, UK), Toronto Research Chemicals (North York, ON) and Cambridge Biosciences UK (Cambridge, UK). A mixed stock solution was prepared in methanol at concentrations of 2, 10, and 25 µg/mL, depending upon the analyte. An internal standard working solution was prepared in 50/50 (v/v) methanol/water at a concentration of 100 ng/mL. Stable isotope labeled internal standards were used for all compounds except in the cases of clonazepam, dehydronorketamine, methedrone, noroxymorphone, and α-pyrrolidinovalerophenone (alpha-PVP) metabolite 1 where stable labeled IS was not readily available. Standards were prepared by diluting the stock solution and spiking dilutions into pooled, blank urine. Quality control (QC) samples were also created by diluting the stock solution and spiking dilutions into pooled, blank urine. All analytes, along with their retention times and calibration ranges are listed in Appendix 1.
Sample extraction can be semi-automated, by utilizing a Hamilton liquid handling robot, enabling sample tracking from the sample tube barcode to the processed sample.
25 µL of urine sample is transferred into a Waters 700 µL Round 96-Well Collection Plate and internal standard working solution is added and thoroughly mixed. Samples are diluted with a distilled water/formic acid solution and mixed prior to injection on the UPLC-MS/MS system.
LC Conditions
|
LC system: |
ACQUITY UPLC I-Class FL IVD |
|
Column(s): |
ACQUITY UPLC BEH C18, 1.7 µm, 2.1 x 100 mm |
|
Column temperature: |
40 °C ± 2 °C alarm |
|
Injection volume: |
20 µL |
|
Mobile phase A: |
Water with 0.1% Formic Acid |
|
Mobile phase B: |
Acetonitrile with 0.1% Formic Acid |
Gradient Table
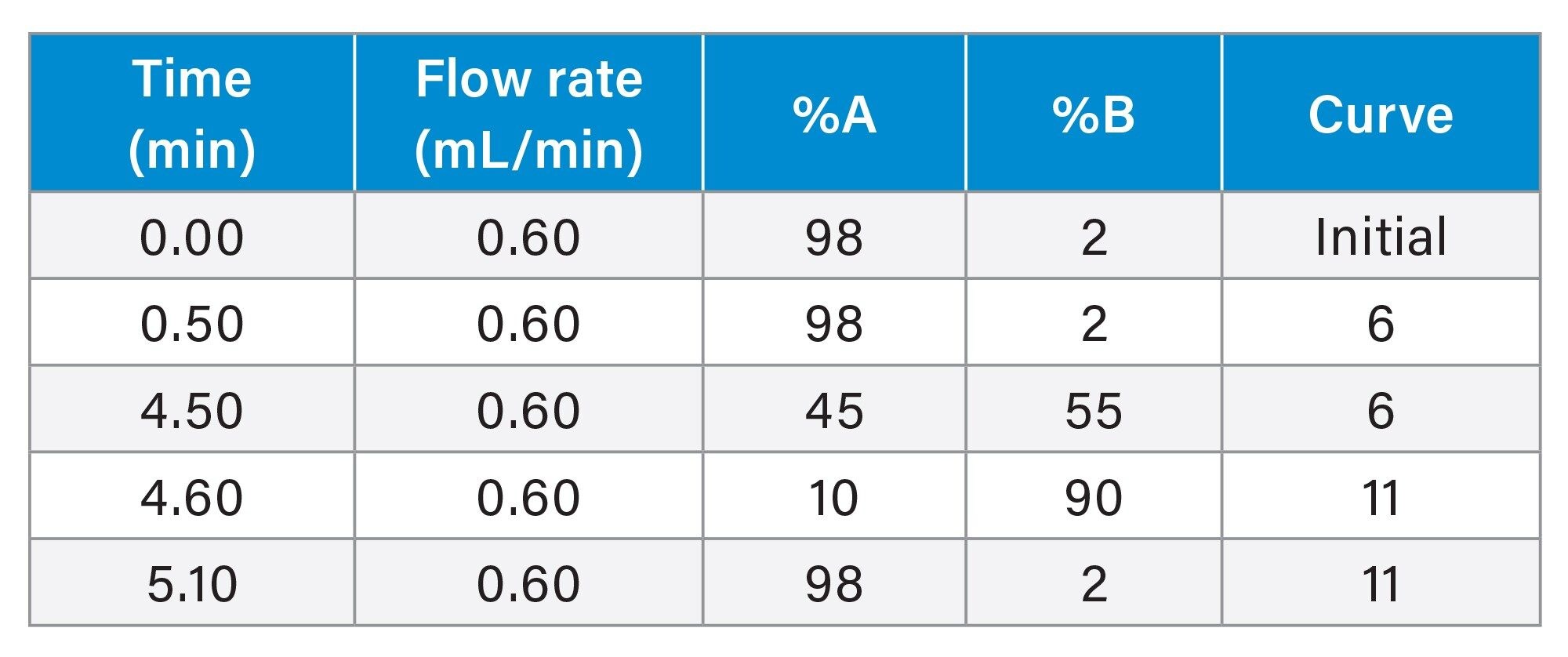
MS Conditions
|
MS system: |
Xevo TQ-S micro IVD |
|
Ionization mode: |
ESI+ |
|
Capillary voltage: |
0.8 kV |
MS method parameters including cone voltage, collision energy and multiple reaction monitoring (MRM) transitions are given in Appendix 1.
Data Management
|
MS Software: |
MassLynx |
|
Informatics: |
TargetLynx™ |
Results and Discussion
Chromatography
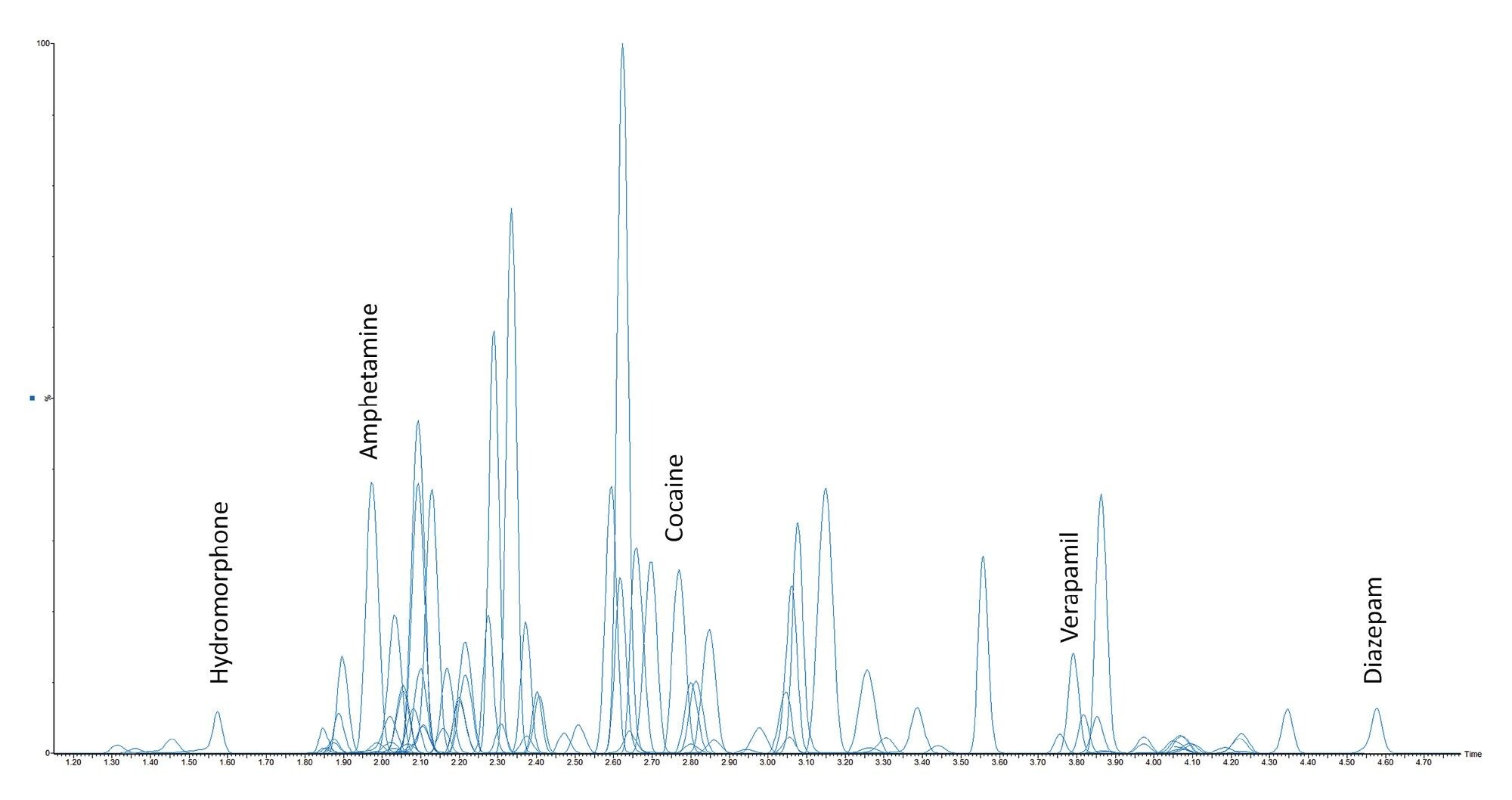
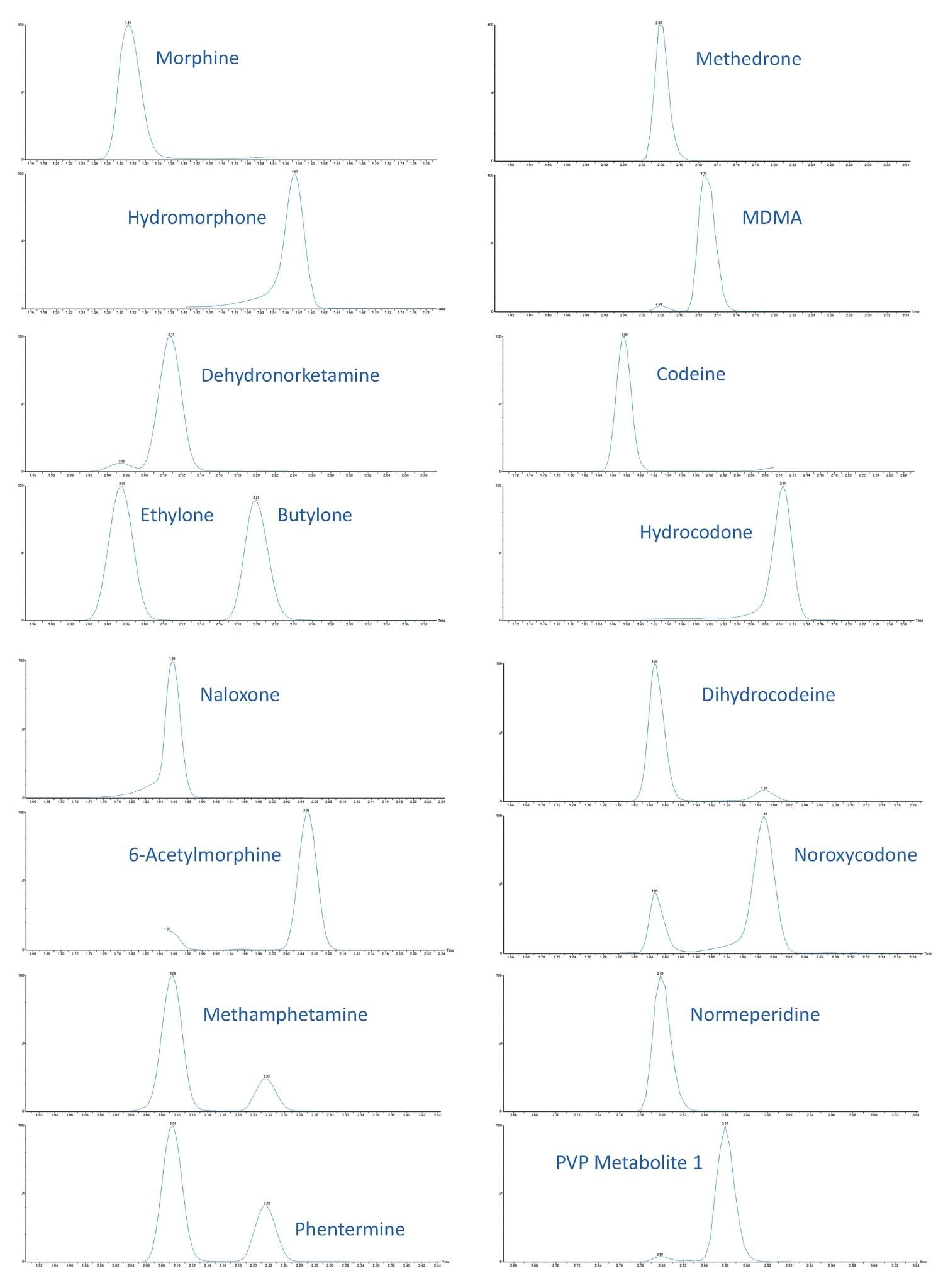
All test compounds are listed in Appendix 1, along with their retention time windows and calibration ranges. Figure 2 shows the overlaid chromatographic separation of all compounds and Figure 3 shows chromatograms from several groups of analytes with the potential to interfere with each other and these are listed below.
|
Morphine and Hydromorphone |
Methedrone and MDMA |
|
Dehydronorketamine, Ethylone, and Butylone |
Codeine and Hydrocodone |
|
Naloxone and 6-Acetylmorphine |
Dihydrocodeine and Noroxycodone |
|
Methamphetamine and Phentermine |
Normeperidine and PVP Metabolite 1 |
In all cases shown, either baseline chromatographic separation is achieved, or selective MRMs are employed and therefore these compounds will not interfere with one another. The UPLC separation was modified slightly when compared to Application Note 720006187 due to the less selective sample clean-up of this dilution method. A temporary hold was employed at the start of the gradient to allow for salts and other matrix components to be diverted to waste before analytes were eluted from the column.
Seven-point calibration lines were extracted in duplicate and analyzed for each analysis run. Calibrators at the higher range were between 25–2,500 ng/mL, calibrators at the mid-range were between 10–1,000 ng/mL and calibrators at the lower range were between 2–200 ng/mL and are shown in more detail in Appendix 1. All calibration lines had a correlation coefficient (r2) of >0.99 and at least 75% of calibration points were within ±15% of their nominal value (±20% at the calibrator 1 level) with the exception of N-desmethylzopiclone, norpropoxyphene, and zopiclone. Further investigation is needed for these compounds to identify whether solubility/stability improvements can be made when preparing the calibrators. N-desmethylzopiclone, norpropoxyphene, and zopiclone were only monitored qualitatively.
QC samples at the higher range were at concentrations of 25, 75, 187.5, and 1875 ng/mL, QC samples at the mid-range were at concentrations of 10, 30, 75, and 750 ng/mL and QC samples at the lower range were at concentrations of 2, 6, 15, and 150 ng/mL and are shown in more detail in Appendix 1. At least 66% of QC samples were within ±15% of their nominal value (±20% at the QC1 level) for all runs.
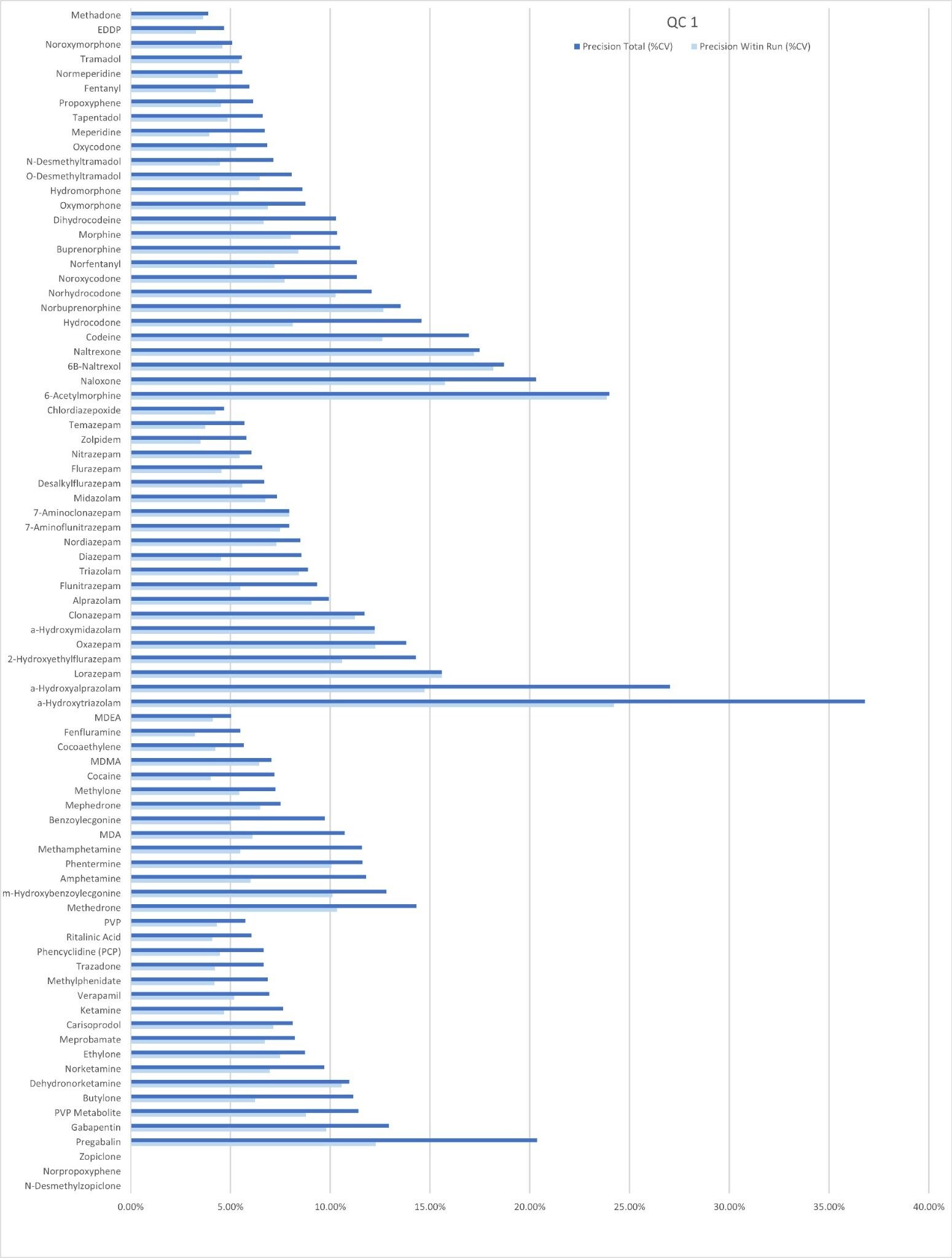
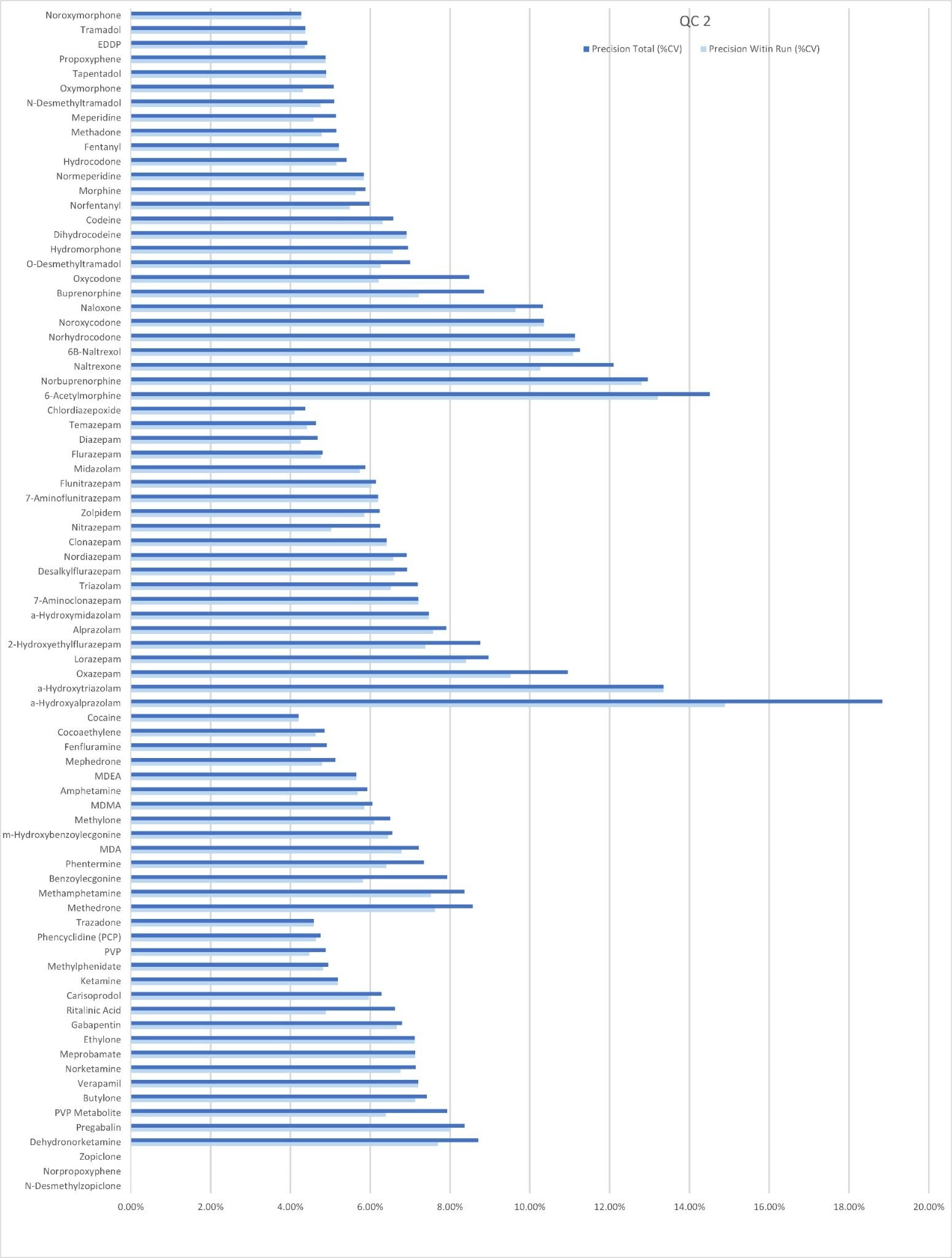
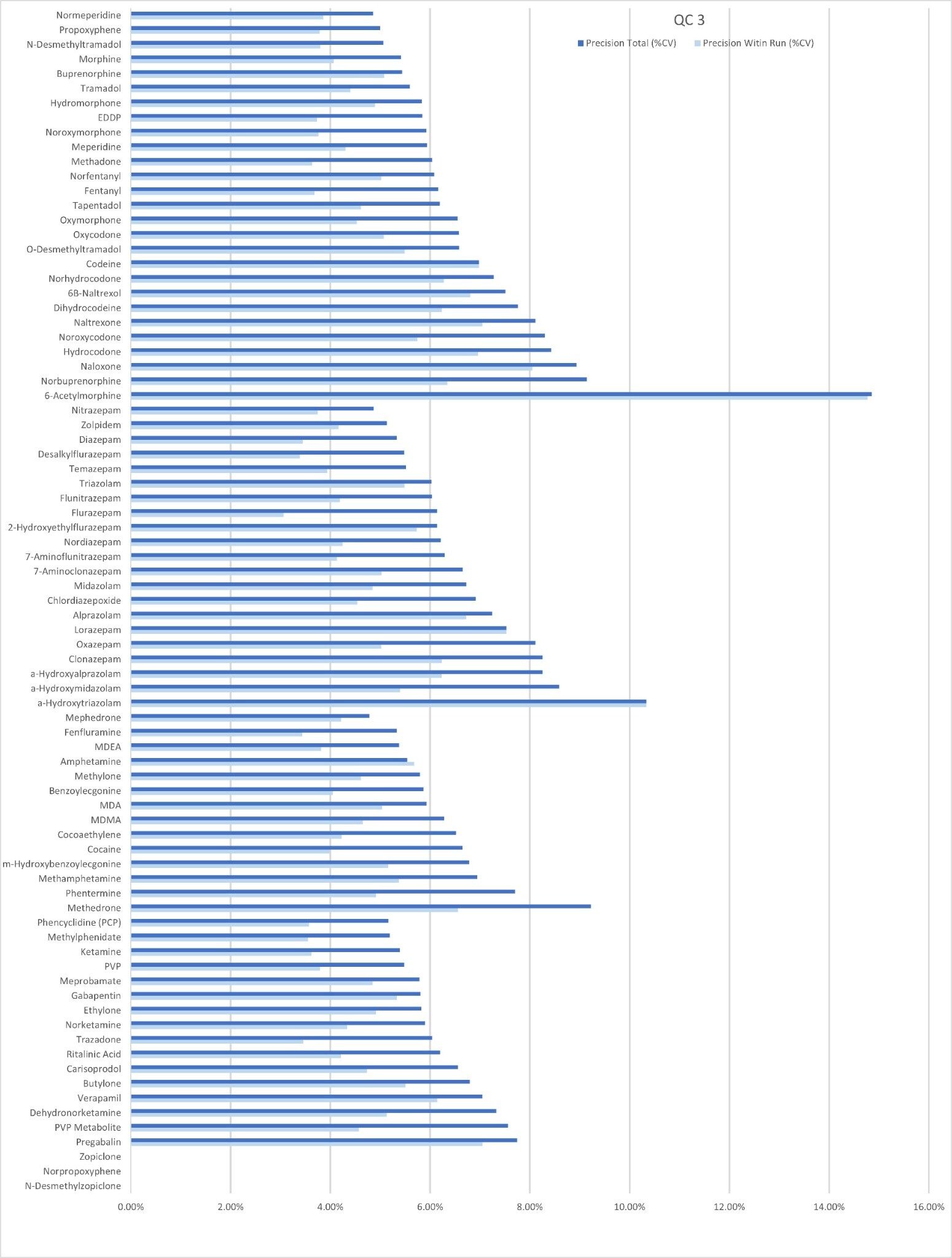
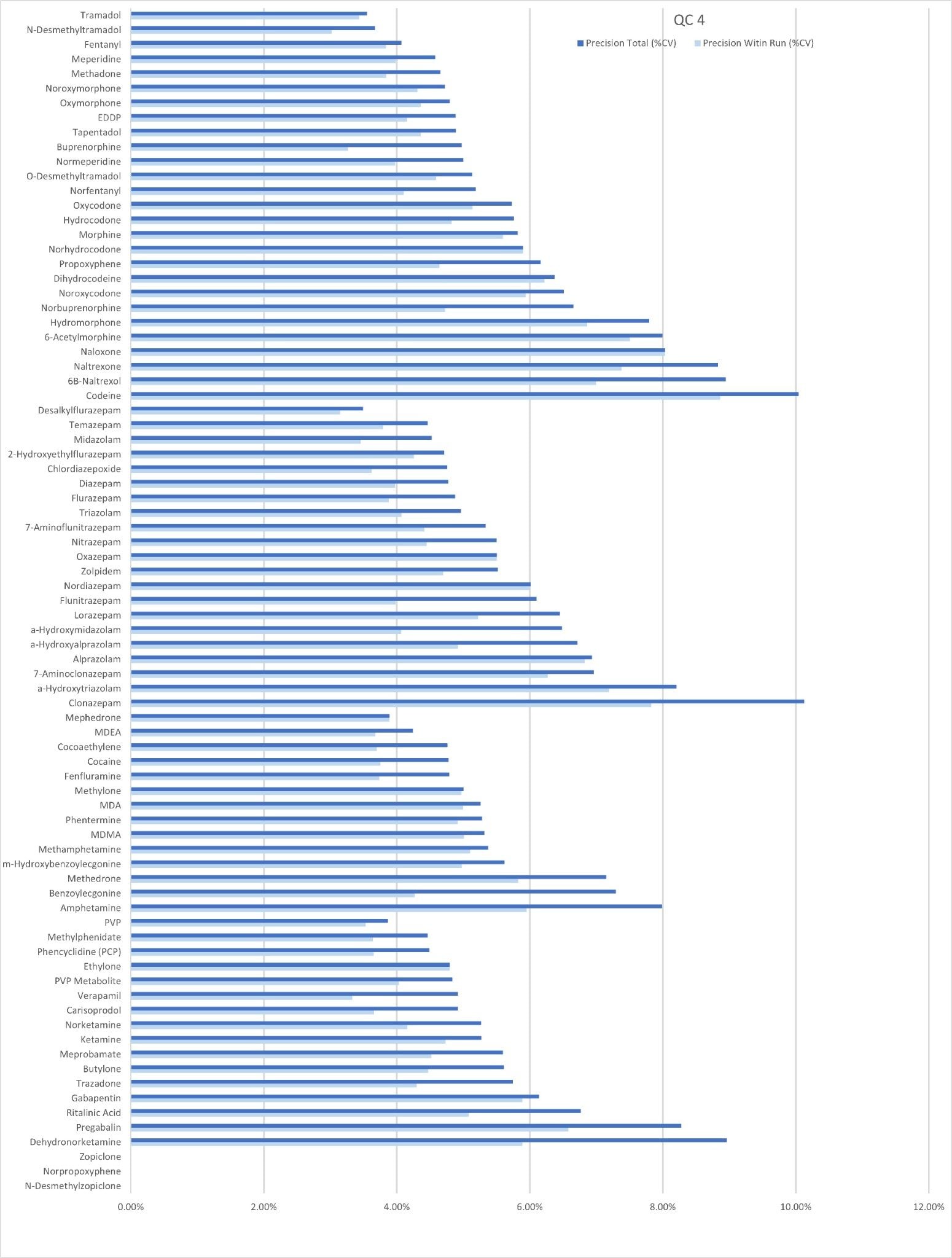
Precision Performance
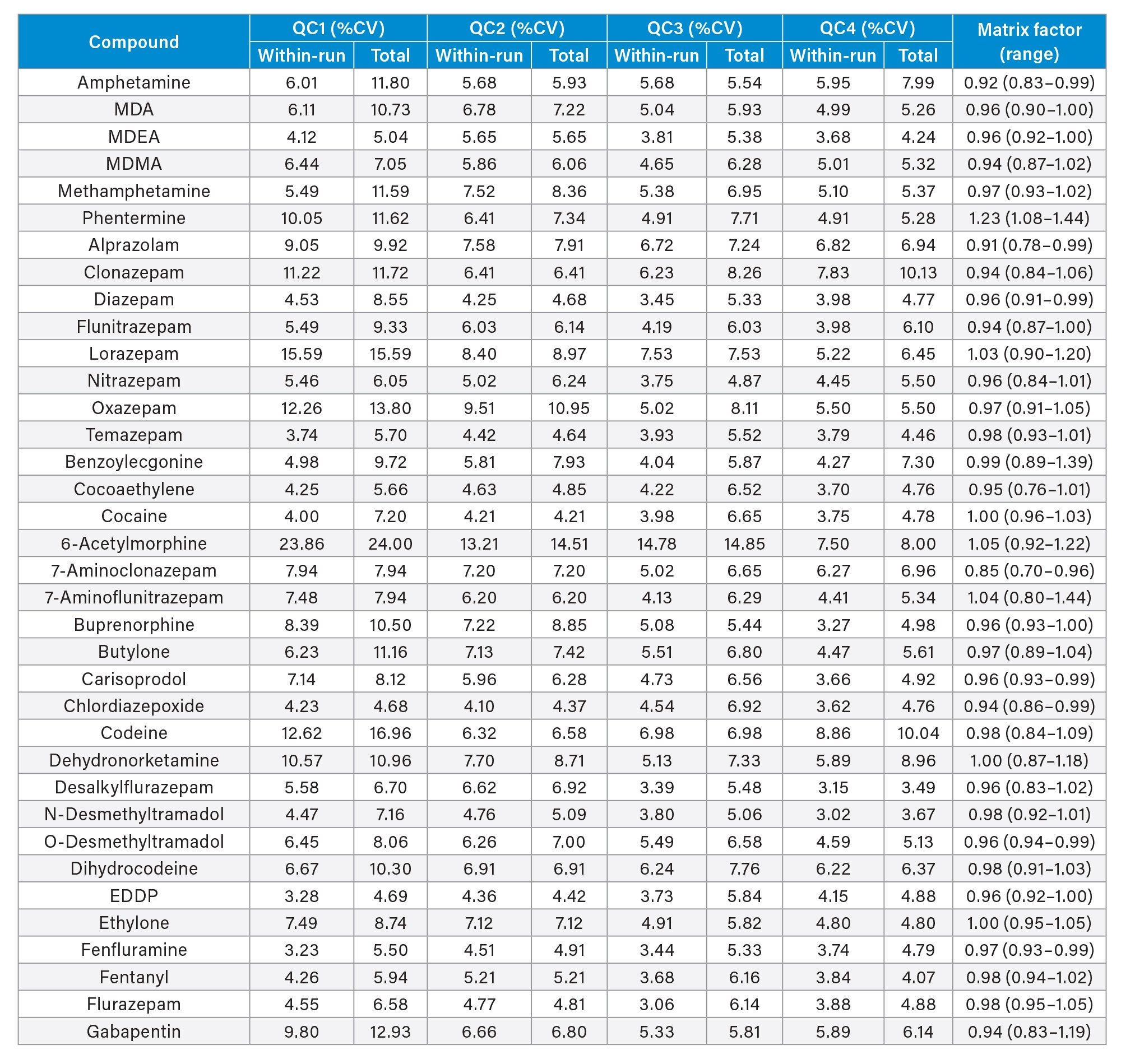
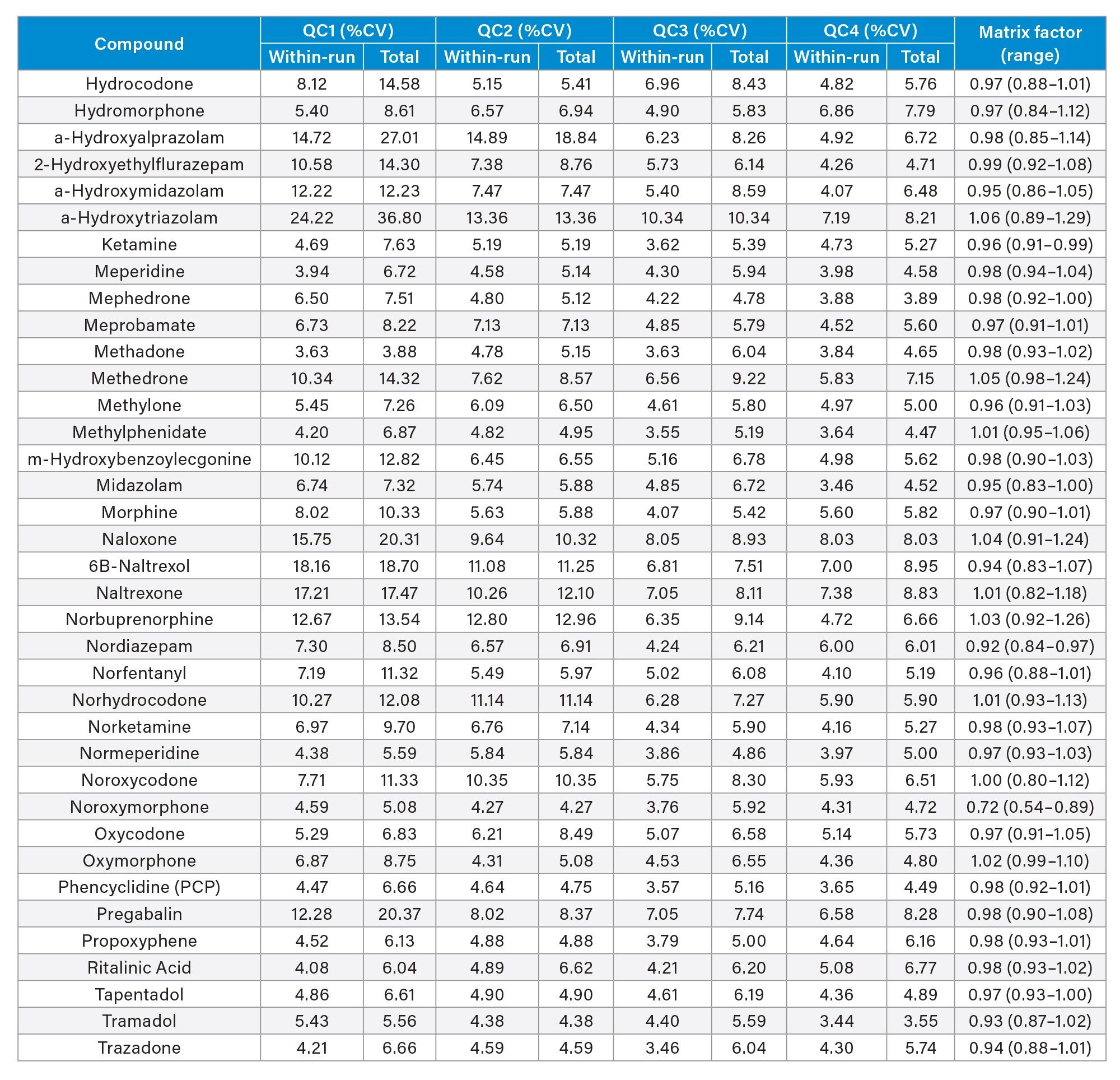
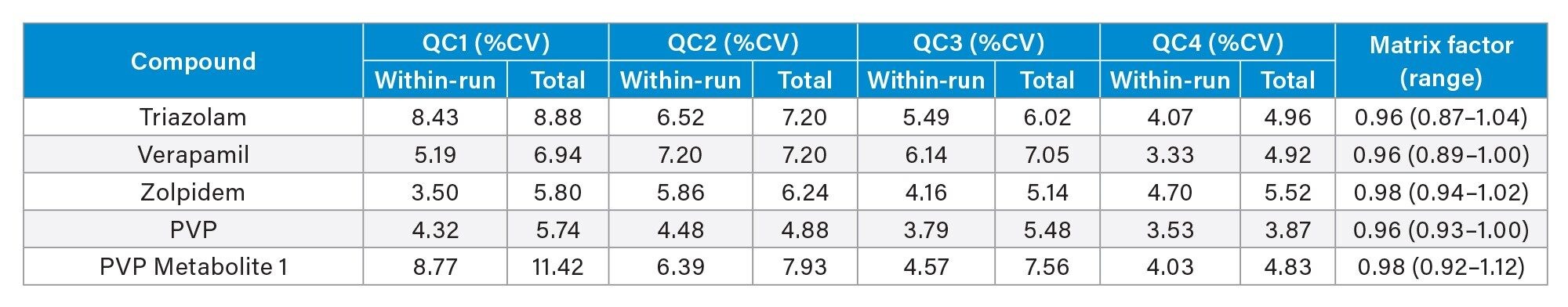
Precision of the method was evaluated by extracting and analyzing five replicates of each QC sample (concentrations above) on each of five occasions (n=25 replicates). Figure 4a to 4d shows a summary of results obtained for each QC level.
Analytical Sensitivity
Ten replicates of low level samples at or below the calibrator 1 concentration were extracted and analyzed on each of four occasions and their %CVs calculated. An acceptance criteria of <20% CV was obtained for the majority of analytes. In the case of lorazepam, 6-acetylmorphine, α-hydroxyalprazolam, α-hydroxytriazolam, naloxone, naltrexone, and pregabalin where this acceptance criteria was not met, it is recommended to employ the sample extraction method described in Waters Application Note: 720006187, if lower concentrations are required.
Carryover
No significant carryover was observed when comparing the mean of six blank matrix samples to those following samples at double the Calibrator 7 concentration for all analytes with the exception of 7-aminoclonazepam and 7-aminoflunitrazepam, which were both found to be at 38.8% of the calibrator 1 sample peak area. Further testing would be required to evaluate the concentration at which carryover becomes insignificant for these analytes.
Matrix Factor
Matrix Factor was between 0.85 and 1.15 (Matrix effects within 15%) when comparing extracted urine samples from six different individuals to control samples in water and post spiked at low and high concentration samples for all analytes with the exception of phentermine and noroxymorphone. A summary of Matrix Factor data for all analytes can be seen in Appendix 2.
Conclusion
This application brief shows an overview for the UPLC-MS/MS analysis of pain management drugs and drugs of abuse for forensic toxicology. Sample preparation has been simplified to a dilute and shoot approach to allow for a fast extraction technique. Coupled with UPLC separation and detection on the Xevo TQ-S micro IVD, a method for the analysis of a large panel of pain management drugs and drugs of abuse has been shown. Results were shown to be precise, with consistent matrix effects and minimal carryover for the majority of analytes within this large panel. A more substantial sample extraction technique is recommended for certain compounds to improve analytical sensitivity such as that shown in Waters Application Note. 720006187.
References
- Danaceau JP, Freeto S, Calton L. A Comprehensive Method for the Analysis of Pain Management Drugs and Drugs of Abuse Incorporating Simplified, Rapid Mixed-Mode SPE with UPLC-MS/MS for Forensic Toxicology. Waters Application Note 720006187. 2019 March.
- Rosano TG, Rumberger JM, Wood M. Matrix Normalization Techniques for Definitive Urine Drug Testing. Journal of Analytical Toxicology, 2021;45:901–912.
Appendix 1
Appendix 2
720007898, June 2023