Methods and Materials
All chemicals were obtained from Sigma-Aldrich, with all compounds having >99.5% purity. All analyses were performed using an Agilent 6890 GC oven fitted with a CTC Combi PAL Autosampler.
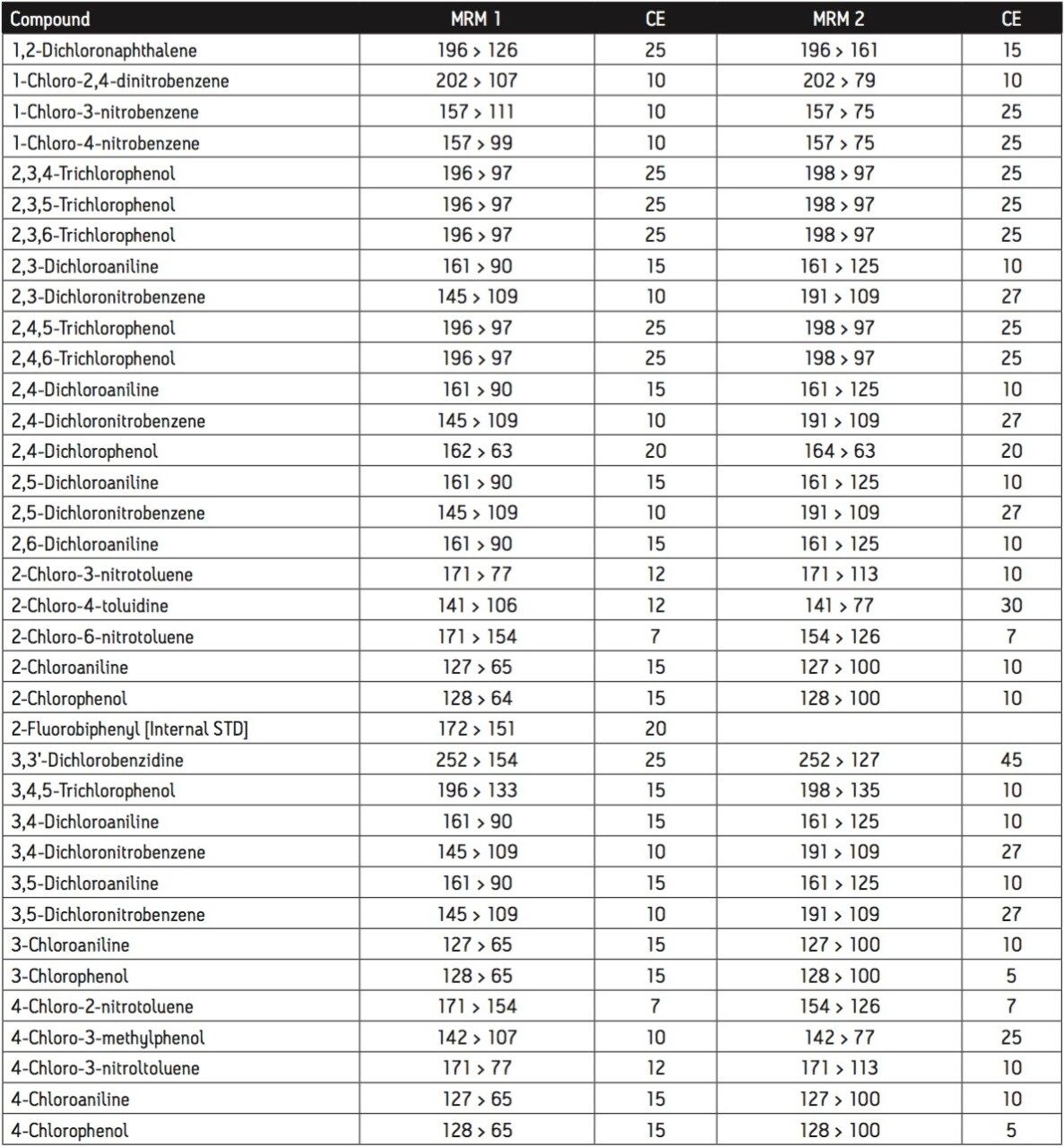
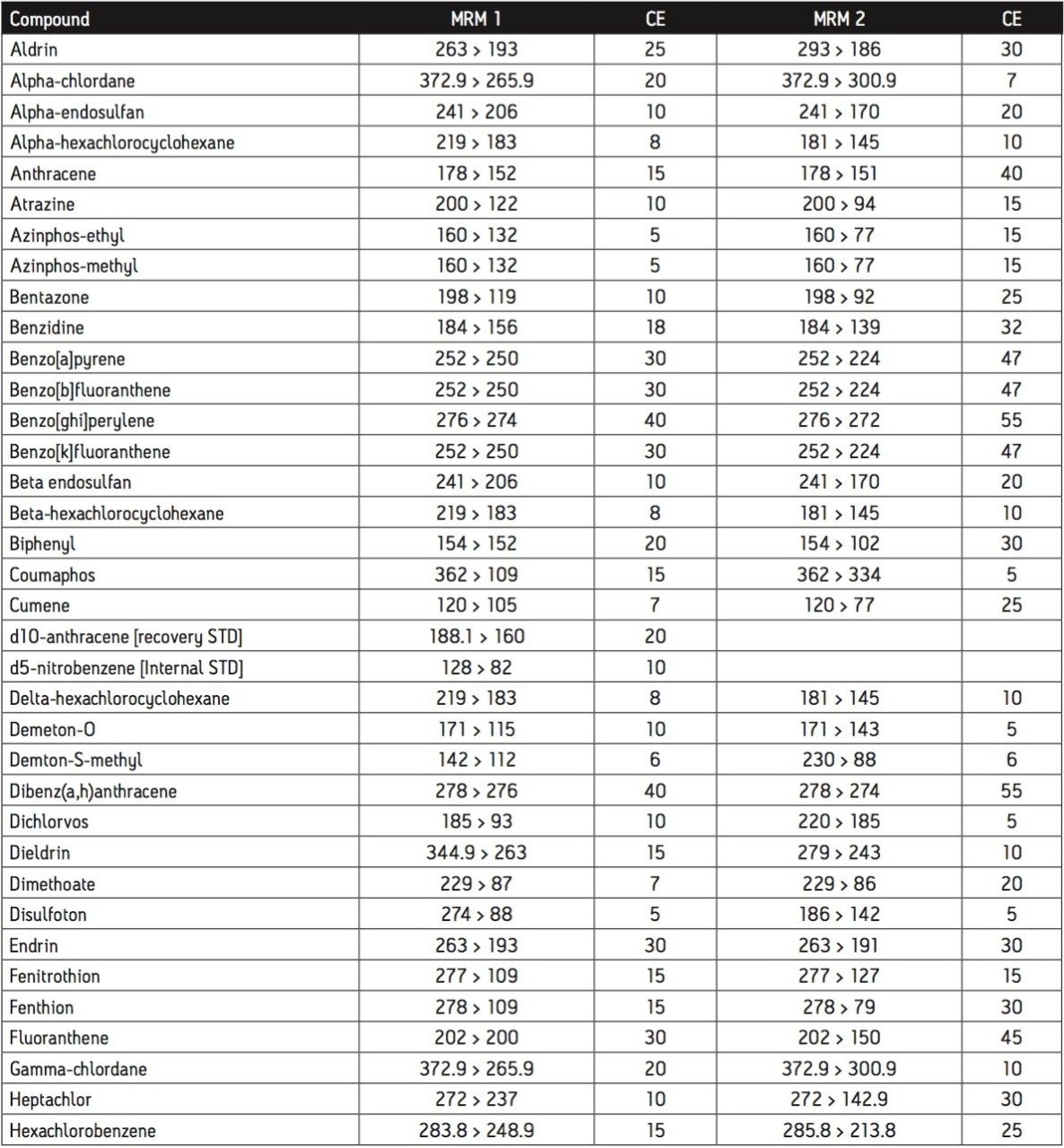
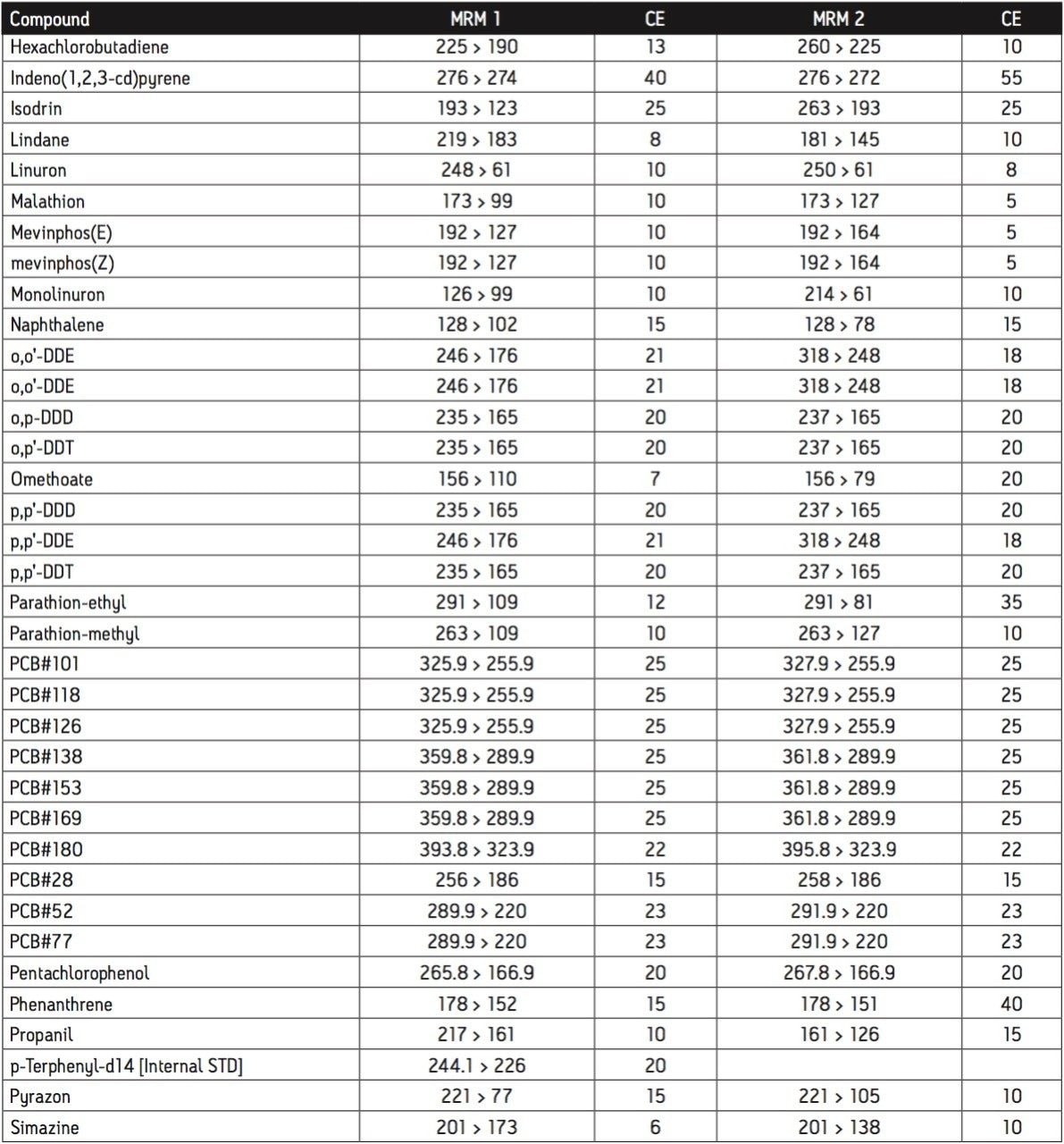
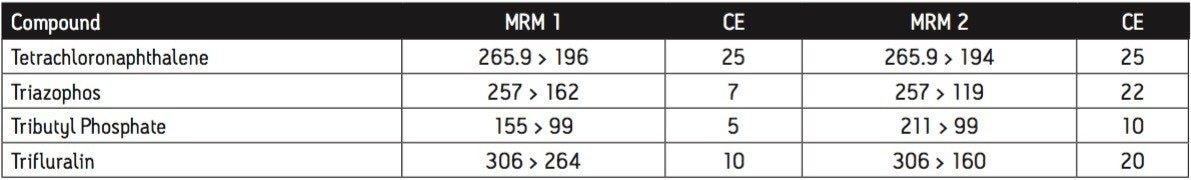
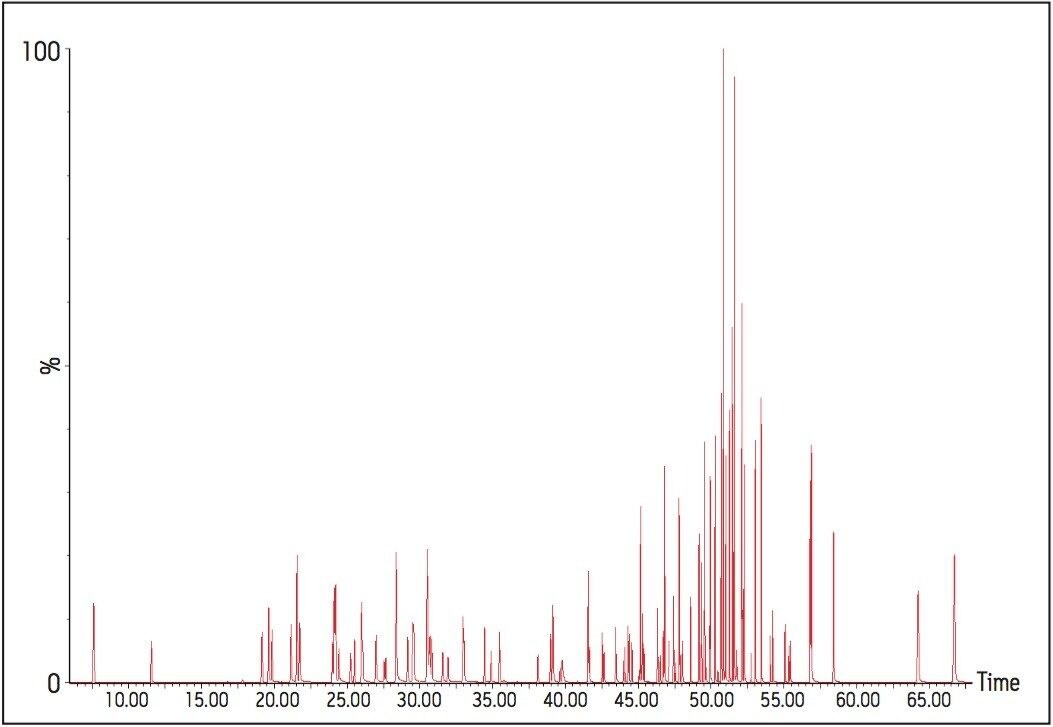
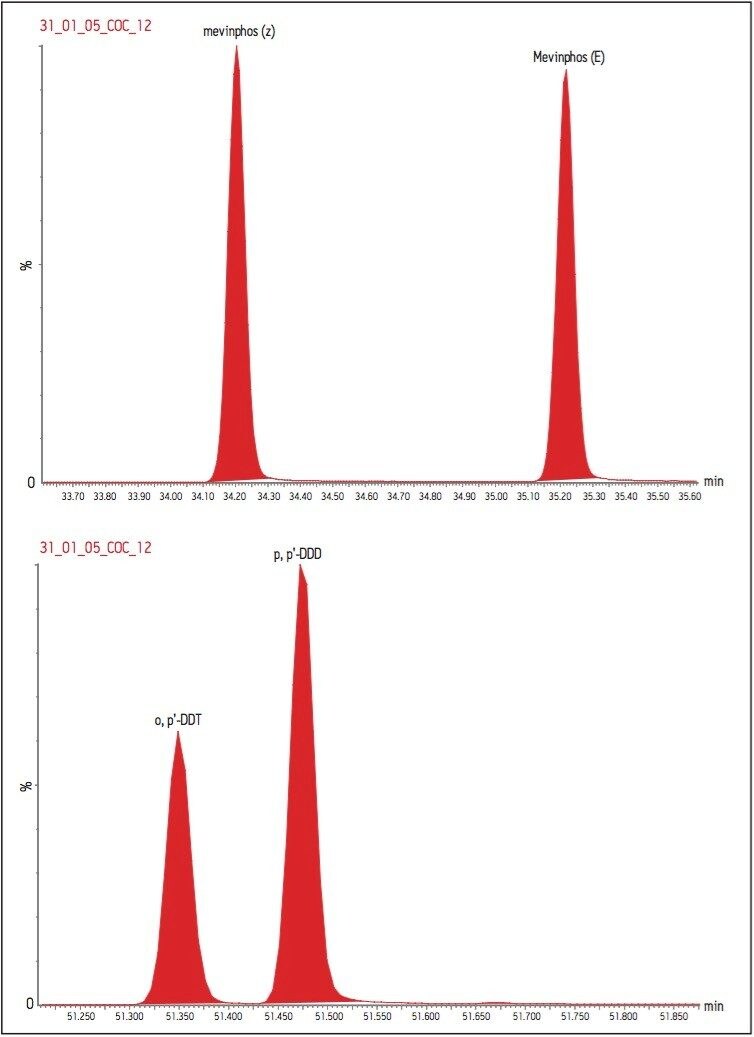
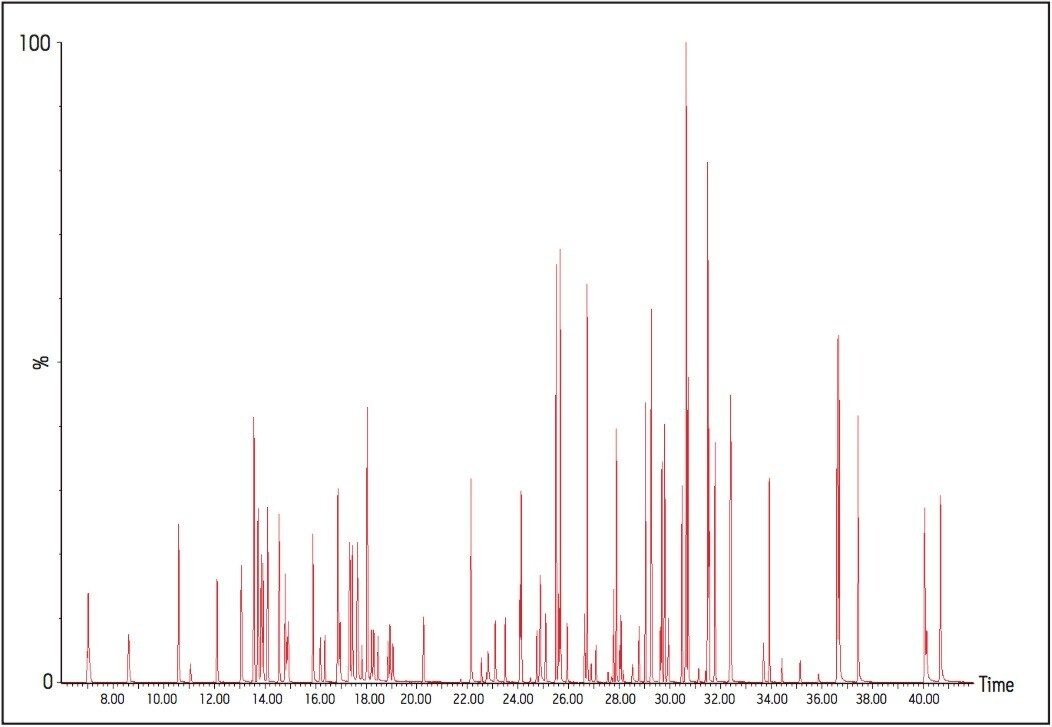
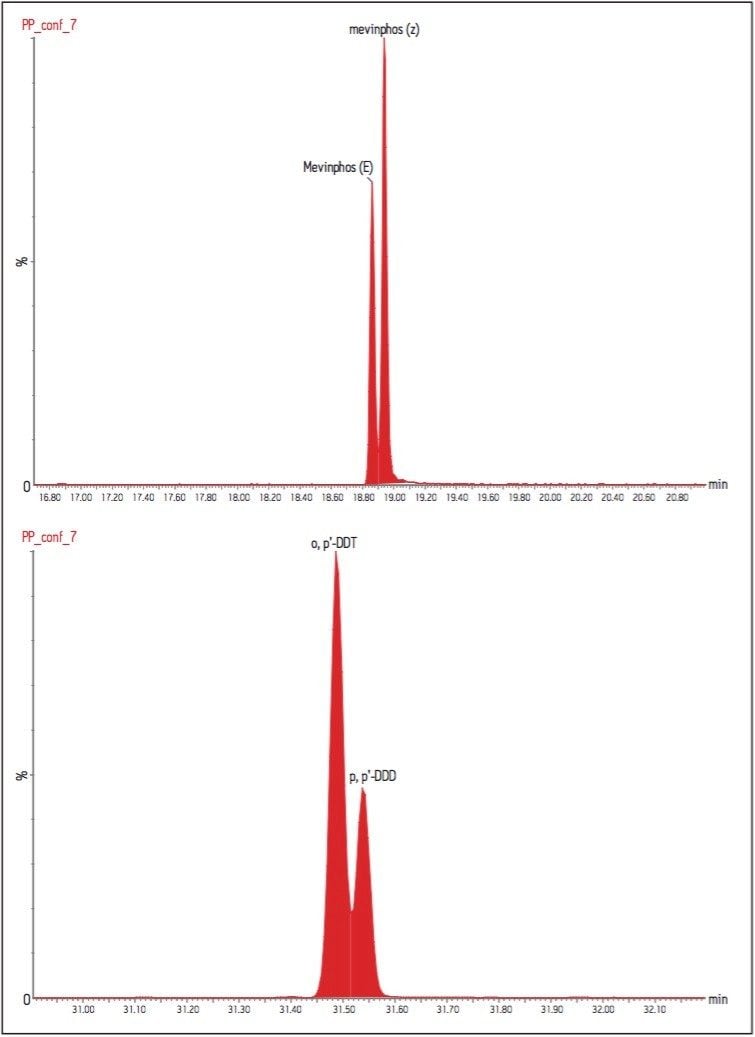
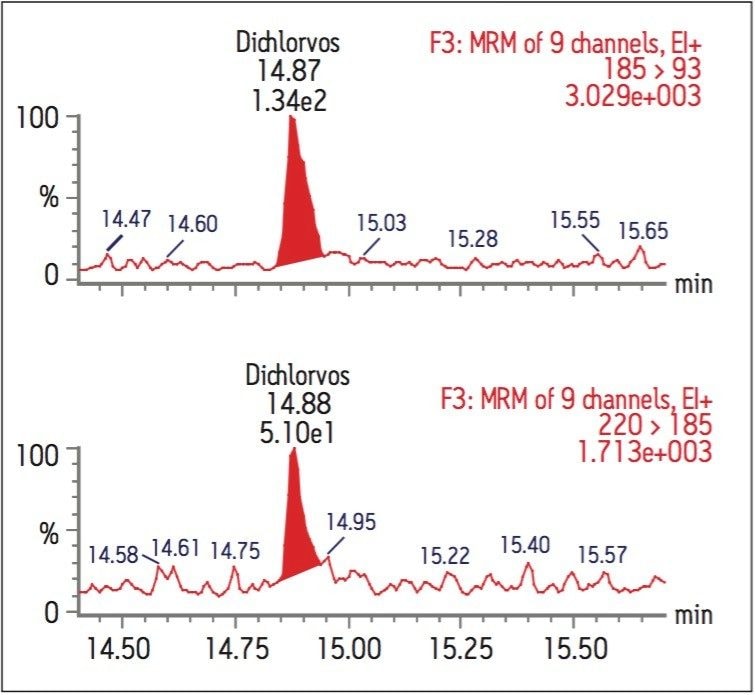
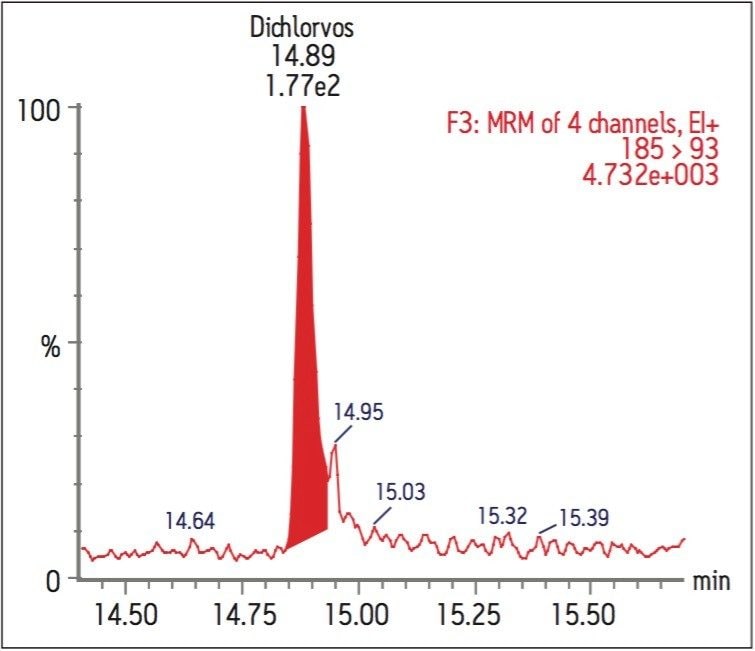
The GC was directly interfaced to a Waters Quattro micro GC Tandem Quadrupole Mass Spectrometer that was operated in the EI+ ion mode. The instrument ion source was operated at 70 eV electron energy, with a source temperature of 180 °C. Three GC columns were evaluated, J&W DB17-ms 30 m 0.25 mm ID, .25 μm df, Restek RTX-5, 40 m 0.18 mm ID, 0.2 μm df and Varian factor four vf5-ms 30 m 0.25 mm ID, 0.25 μm df. Injections were made using both pulsed splitless and cool on column (COC) injections, with a 2 m 0.53 mm ID retention gap fitted for COC injections. All compounds were acquired in full scan and daughter scanning acquisition modes, with the results used to optimize at least two MRM transitions per compound. Internal and recovery standards had one MRM transition optimized. MRM analysis was performed using a single transition per compound, where confirmation is based upon one MRM transition plus the retention time, and also using two MRM transitions per compound, where the strictest EU confirmatory criteria are satisfied. The difference in sensitivity between the two approaches was compared. The three GC Columns were assessed for chromatographic resolution of critical pairs of co-eluting peaks, overall run time, and sensitivity of active components. All standards were prepared from >99.5% purity solids dissolved in dichloromethane (DCM), with a mixed standard being prepared at a concentration of 5 ng/L in DCM, and also acetone (for spiking purposes).
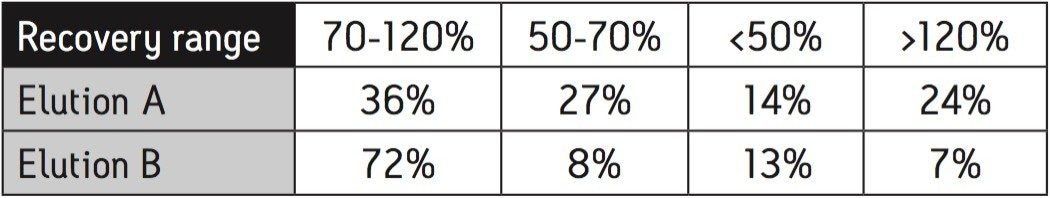
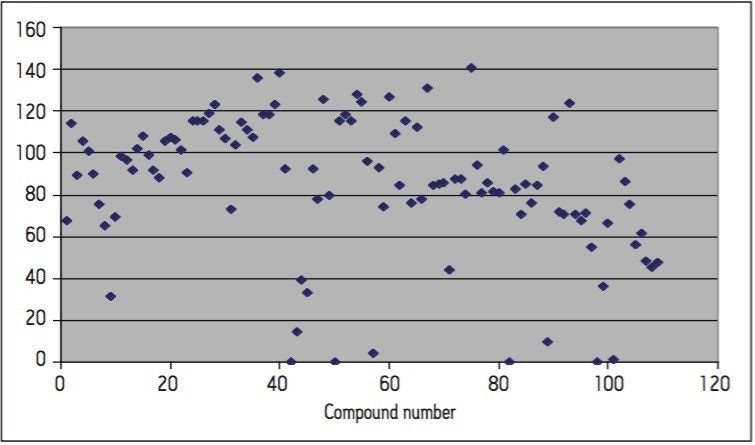
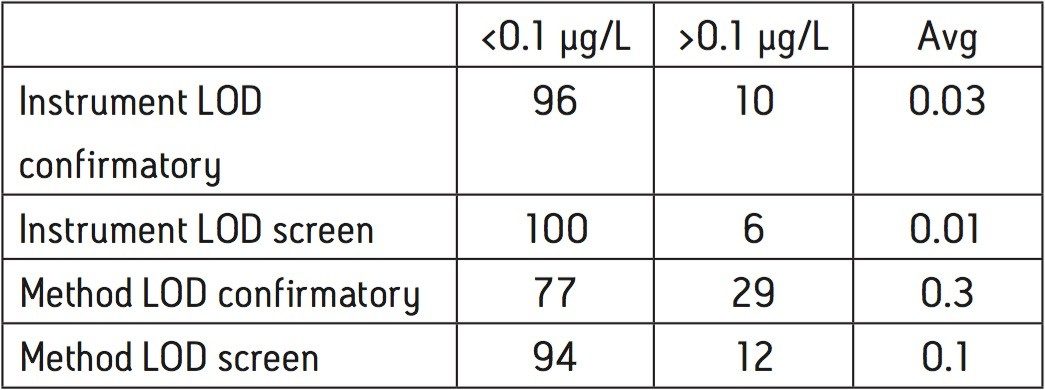
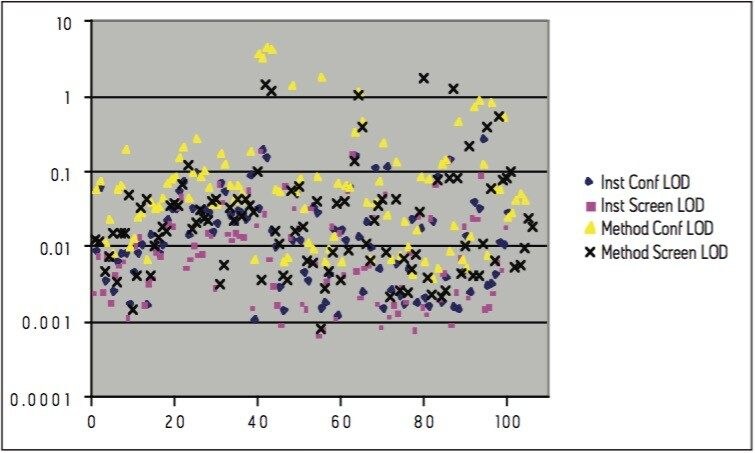
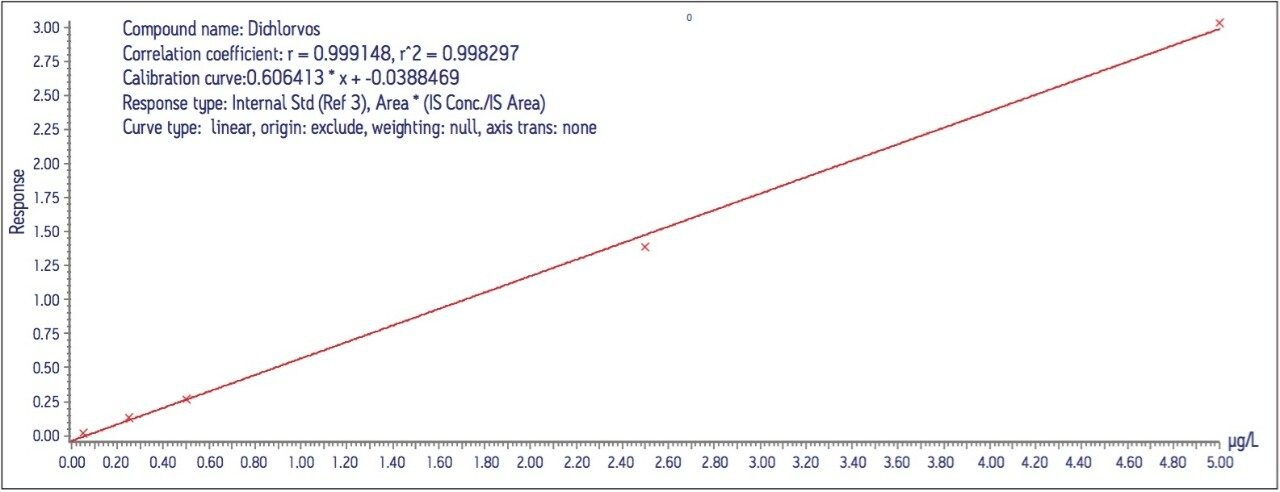
Calibration curves were acquired over the concentration range of 0.05 to 5 μg/L. Extraction and clean-up were performed using Waters Oasis HLB 3cc, 60 mg SPE cartridges. 200 mL of each filtered water sample was spiked with an internal standard mixture containing d5-nitrophenol, 2-fluorobiphenyl and p-terphenyl-d14 at a level of 500 ng for each component. The water was adjusted to pH4 using 1 N HCl solution. The SPE cartridges were conditioned with 6 mL DCM, 6 mL acetonitrile and 6 mL of water at a flow rate of 3 mL/min. The water samples were then loaded at a flow rate of ca 6 mL/min. After sample loading was completed, the cartridges were washed with 1 mL water. The cartridges were then dried under a flow of nitrogen (ca 1 mL/min) for 20 mins, followed by final elution with either A. 2.5 mL DCM/ACN (4:1), 5 mL DCM; or B. 5 mL DCM. After elution, the extract was adjusted to a volume of ca 0.5 ml under a stream of dry nitrogen at ambient temperature, followed by the addition of 500 ng of d10-anthracene as a recovery standard. Drinking and canal water samples were spiked with the analytes at concentrations of 0.5 μg/L and 5 μg/L prior to extraction for recovery tests.
The GC temperature ramps employed were:
30 m DB17-ms
40 °C/1 min, 3 °C/min to 160 °C, 7 °C/min to 240 °C, 15 °C/min to 305 °C, hold 15 mins. 1 mL/min He flow
40m RTX5
40 ° C/1 min, 3 °C/min to 160 °C, 7 °C/min to 240 °C, 15 °C/min to 310 °C, hold 15 mins. 0.7 ml/min He flow
30 m vf5-ms
40 °C/0.8 min, 6 °C/min to 160 °C, 8 °C/min to 310 °C, hold 2 mins. 0.9 mL/min He flow
All injections in pulsed splitless mode were made with an injection temperature of 250 °C, using a double gooseneck 4 mm ID liner and 1 μL injection volume. The injections were made with a 1 min 110 kPa pulse, a purge time of 1 minute and a purge flow of 70 mL/min.
Cool on column injections were made in track oven mode.
Data were acquired with Waters MassLynx Software and processed with Waters TargetLynx Application Manager.