The pharmaceutical industry struggles with solving method transfer challenges. Implementation of UPLC increases overall profitability and as a result, pharmaceutical companies are transferring their legacy USP HPLC methodology to UPLC to increase efficiency within the QC environment. Therefore, simplified approaches supplemented with proven examples are essential to assisting their method translation goals with experience.
The compendial method for lamotrigine is a good representation of typical conditions described in other USP HPLC methodology. Lamotrigine is an anticonvulsant drug approved by the FDA and marketed by GSK as Lamictal primarily to treat seizures of specific diagnosis experienced by patients with epilepsy. For the treatment of bipolar depression, one-third of surveyed psychiatrists identify lamotrigine as the therapy with the greatest overall efficacy when compared to other currently available treatments.1 Since the patent expiration in 2008, a report published by Allbusiness.com titled “GlaxoSmithKline (GSK) Q1- 2010” identified competition from generic pharmaceuticals accounted for a 26% y-o-y decrease of Lamictal sales since 2008.2
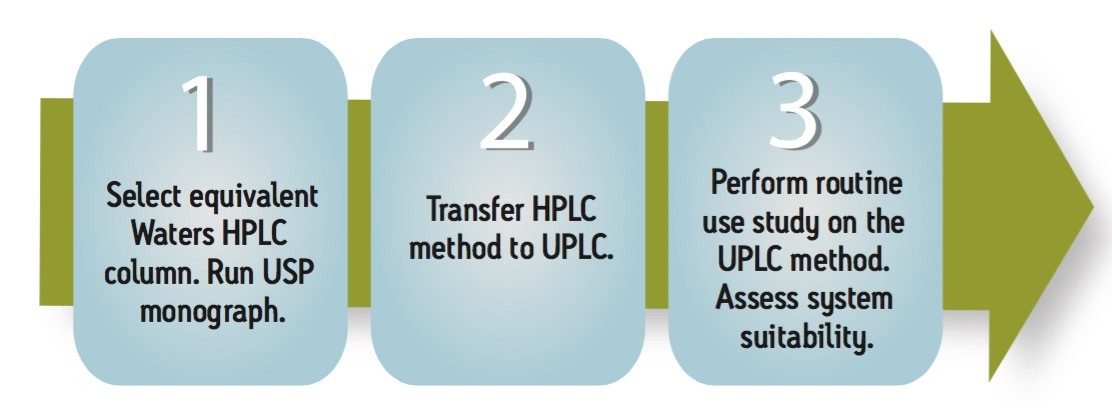
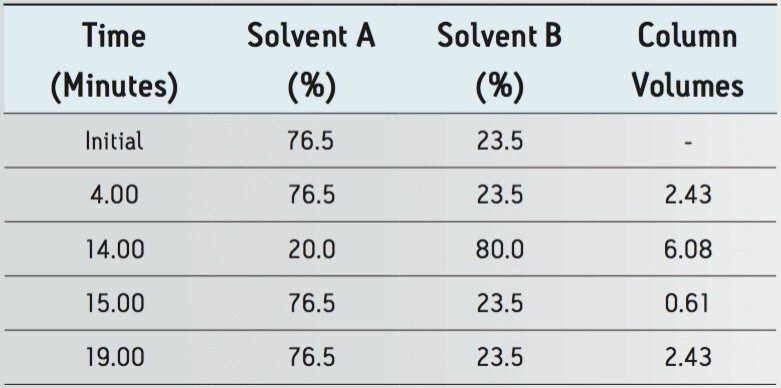
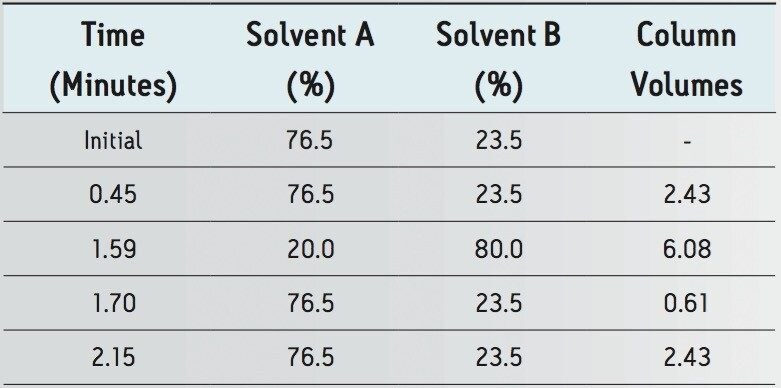
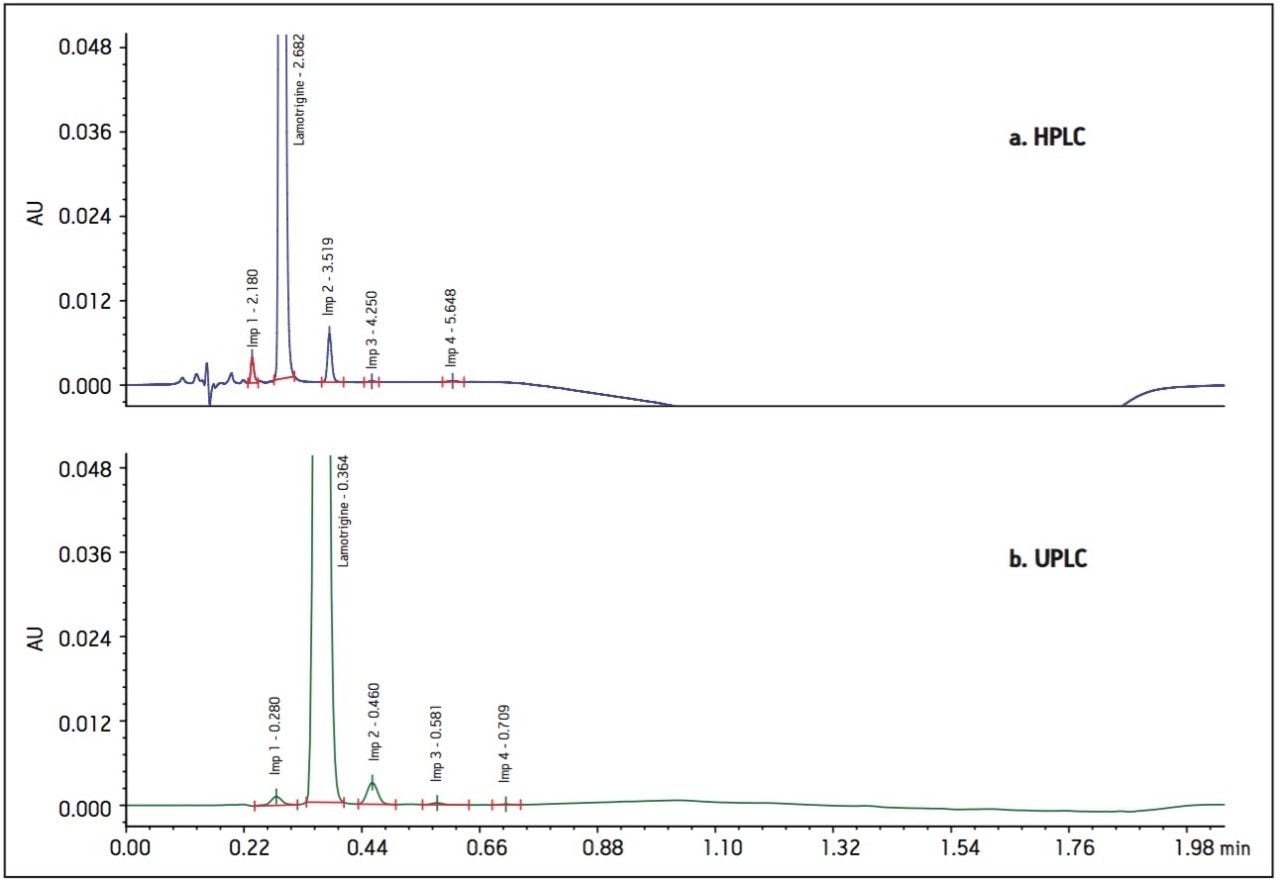
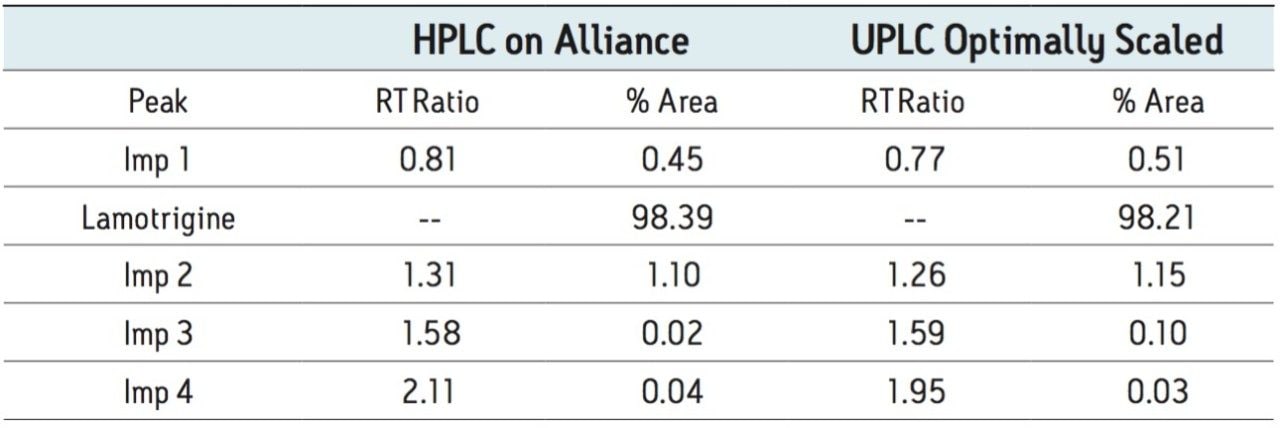
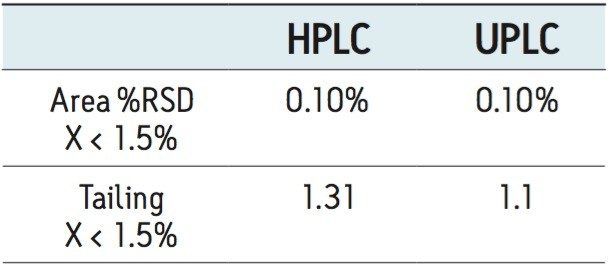
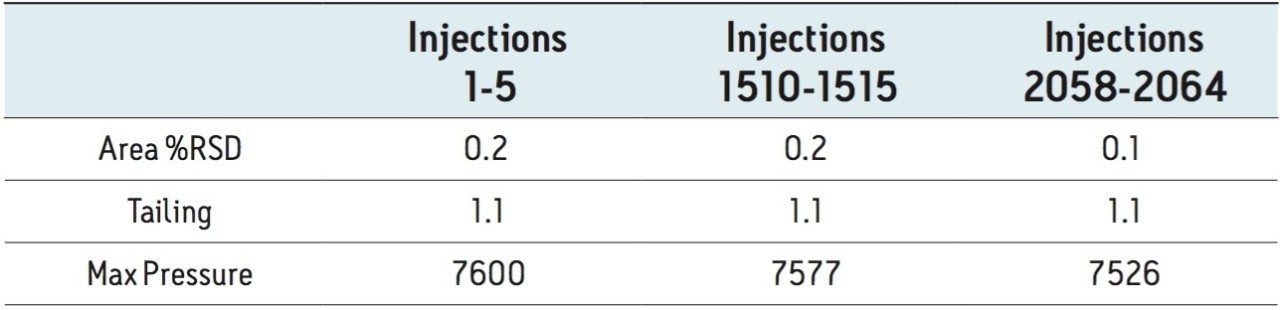
The goal of transferring the USP Lamotrigine HPLC methodology to UPLC was achieved by following the workflow illustrated in figure 1 derived from the strategy described in a previous application note.3 A drug formulation analysis procedure was developed based on the drug substance methodology found in the USP for lamotrigine.4 Method feasibility was determined through routine use studies designed to assess applicability within quality control laboratories.