Method Development for Preparative Purification of Long Oligonucleotides
Alexander Todorova, Stella VerkhnyatSkayaa, Imke Nijdama, Martin Gilarb, Mandana Fasthb
a ProQR Therapeutics, Leiden, The Netherlands
b Waters Corporation, Milford MA, United States
b Waters Corporation, Stockholm, Sweden
Published on October 10, 2025
Abstract
The development of efficient analytical methods for the purification of synthetic oligonucleotides is critical for ensuring drug safety and efficacy. In this work, the systematic evaluation of multiple liquid chromatography (LC) columns and mobile phases to optimize the purification of a 60-mer, 2’-O-methyl and 2′-hydroxyl modified oligonucleotide from process-related impurities is described. A range of stationary phases and elution conditions were screened, with the optimal separation achieved using a batch tested wide-pore XBridge™ Premier Oligonucleotide BEH™ C18, 300 Å, OBD™ Prep Column in combination with a hydrophobic alkylamine ion-pairing reagent. This approach enabled superior resolution of the target oligonucleotide from structurally similar impurities, thereby facilitating high-purity recovery at a lab scale. The results presented here provide valuable insights into optimizing oligonucleotide purification strategies for enhanced process development.
Benefits
- Batch tested and selected XBridge Premier Oligonucleotide BEH C18, 300 Å Column provided predictable, low secondary interaction separations in ion-pair reversed phase chromatography (IP-RP) mode.
- Hardware components with MaxPeak™ HPS Technology minimize non-specific adsorption of oligonucleotides and improve recovery of early eluting sample components.
- Construction with a low adsorption, corrosion resistant hardware design to provide long-lived performance of columns.
- The XBridge Premier Oligonucleotide BEH C18, OBD Preparative Column provides direct and predictable scale-up from pre-purification assessments using MaxPeak Premier Analytical Columns.
- Enhanced retention and selectivity upon optimizing mobile phases.
Introduction
Therapeutic oligonucleotides, such as antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs), require stringent purity specifications to ensure their safety and efficacy. Process-related impurities, including truncated, deleted, and chemically modified sequences, pose significant challenges in analytical method development and purification. These impurities can arise from incomplete synthesis, degradation, or side reactions during manufacturing and must be effectively separated to meet regulatory standards.
Achieving high-purity oligonucleotide isolation is particularly complex for longer sequences, such as the 60- and 70-mer oligonucleotides with 2′-OMe and 2′-OH modifications that were investigated in this study. The presence of multiple hydrophobic modifications can impact chromatographic behavior, making the analysis and purification difficult. Inefficient separation may lead to co-elution of impurities, which can compromise the therapeutic performance and introduce potential safety risks.
To address these challenges, it was systematically screened with various LC stationary phases and mobile phase compositions to optimize impurity separation and improve purification efficiency. The wide-pore XBridge Premier Oligonucleotide BEH C18, 300 Å Sorbent (stationary phase) combined with a hydrophobic alkylamine as the ion-pairing reagent (mobile phase) was identified as the optimal composition for achieving high-resolution purification. This method provided enhanced peak resolution, reduced secondary interactions, and improved oligonucleotide recovery.
The selection of chromatographic resins for the stationary phase is critical to achieving optimal oligonucleotide separation. Resins with very low levels of ionic interactions help minimize secondary interactions that could interfere with separation efficiency. Additionally, mechanical stability at high temperatures and pH resistance are essential for maintaining consistent performance under varying purification conditions. The pore size of the resin must be carefully chosen to accommodate the length of the oligonucleotide, ensuring efficient mass transfer and minimizing peak broadening. For long oligonucleotides such as the 60-mer investigated here, a sufficiently large pore size, such as 300 Å, facilitates better mass transfer by allowing oligonucleotides to enter and exit the stationary phase more efficiently, reducing peak broadening and improving resolution.
Furthermore, the choice of alkylamines as ion-pairing reagents plays a significant role in oligonucleotide purification. Depending on the oligonucleotide’s length, size, and sequence composition, different alkylamines can provide varying degrees of retention and resolution. More hydrophobic alkylamines, as used in this study, enhance oligonucleotide retention and resolution, particularly for sequences with increased hydrophobic modifications. To selectively separate oligos based on their length, more hydrophobic alkylamines such as HA (hexylamine) can be considered. TEAA (triethylammonium acetate) or HAA (hexylammonium acetate) are volatile and can be removed from a collected fraction by evaporation or lyophilization. Triethylammonium bicarbonate presents yet another interesting volatile mobile phase option.
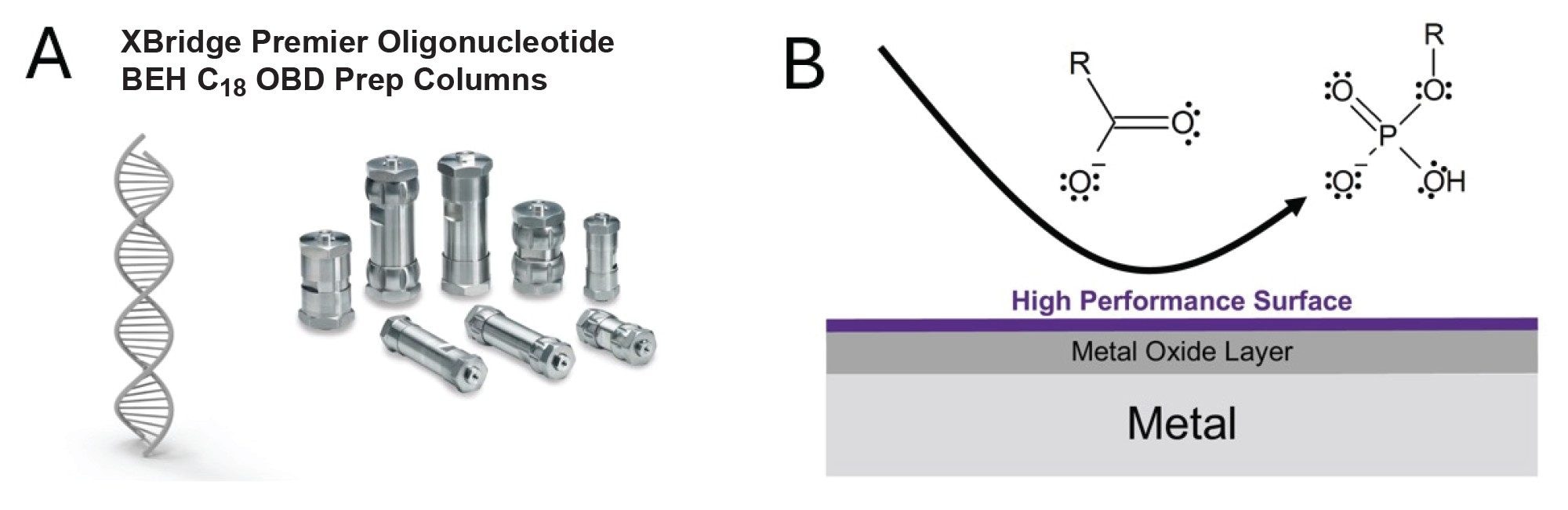
This work highlights the importance of systematic chromatographic method development for oligonucleotide purification and offers a practical approach for improving impurity removal in lab-scale processing. The findings presented here contribute to advancing purification strategies critical for regulatory compliance and therapeutic success.
Experimental
Sample Preparation
The oligonucleotides were prepared using the Mermade™ 12 synthesizer following standard procedures for synthesis, cleavage and deprotection (C&D) and TBS deprotection2. The samples were then split into two (V:V) before lyophilizing. The lyophilized powder was dissolved in 5% ACN, 100 mM hexylammonium acetate (HAA), pH 7.0 water to a concentration of 20 mg/mL or 5% ACN, 100 mM triethylamine acetate (TEAA), pH 7.0 water to a concentration of 20 mg/mL and stored at -20 °C. Before every purification, 1 mL of the stock solution was transferred to a 2 mL HPLC Vial.
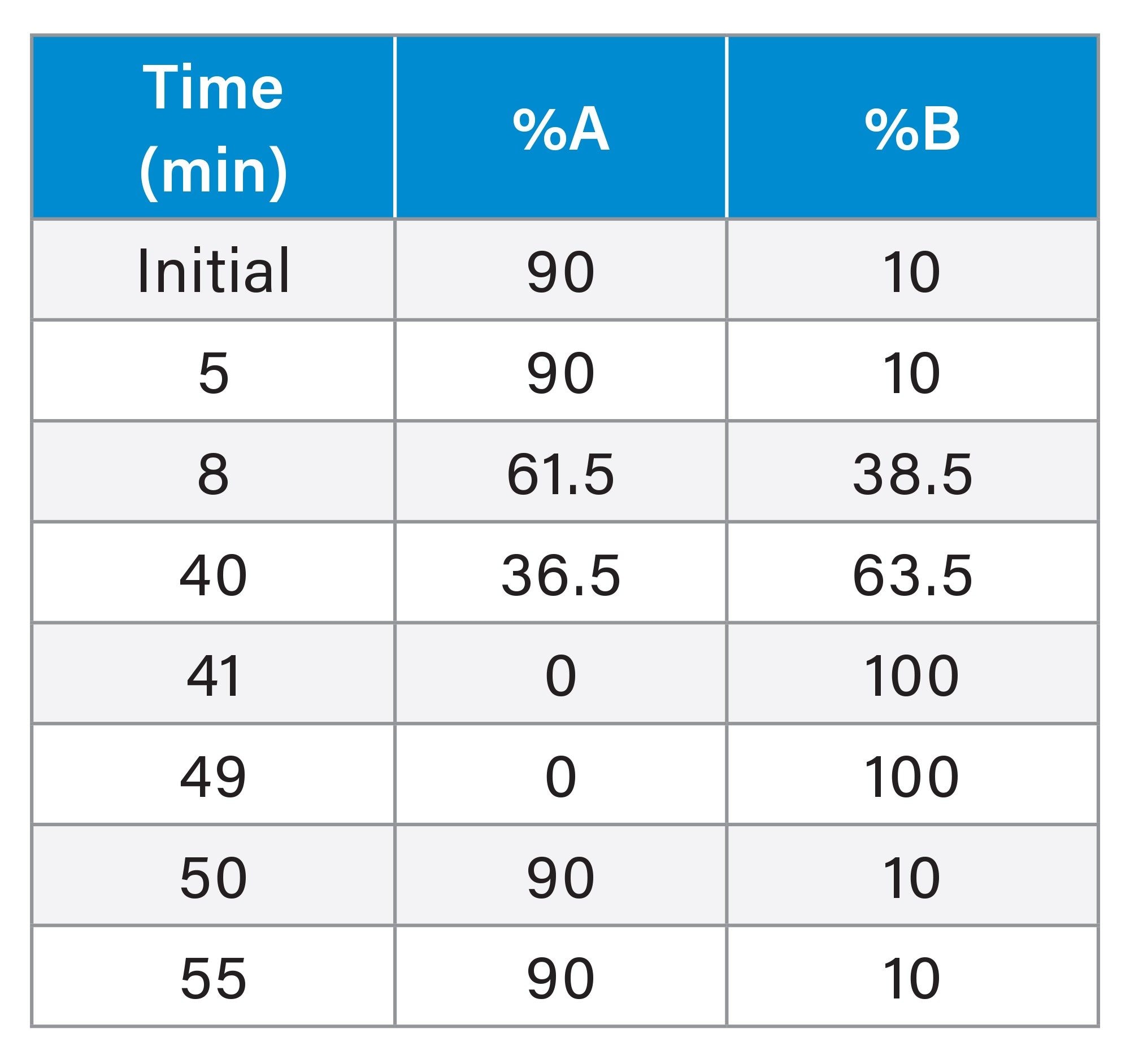
Method Development of Ion-Pair Reversed-Phase Liquid Chromatography (IP-RP LC)
|
Parameter |
Value |
|
LC system: |
Thermo Scientific UltiMate™ 3000 LC System |
|
Column: |
XBridge Premier Oligonucleotide BEH C18 Column, 300 Å, 2.5 µm, 4.6 X 100 mm (p/n: 186010546) |
|
Buffer: |
A1: 5% ACN, 100 mM TEAA, pH 7.0 water B1: 80% ACN, 100 mM TEAA, pH 7.0 water A2: 5% ACN, 100 mM HAA, pH 7.0 water B2: 80% ACN, 100 mM HAA, pH 7.0 water |
|
Flow rate: |
1 mL/min |
|
Injection volume: |
5 µL (in corresponding buffer A) |
|
Mass load: |
5 nmol |
|
Column temperature: |
60 °C |
|
Detection: |
Dionex UltiMate™ 300 RS Diode ARRAY Detector, 260 nm |
Table 1. Machine parameters for the development of the IP-RPLC method.
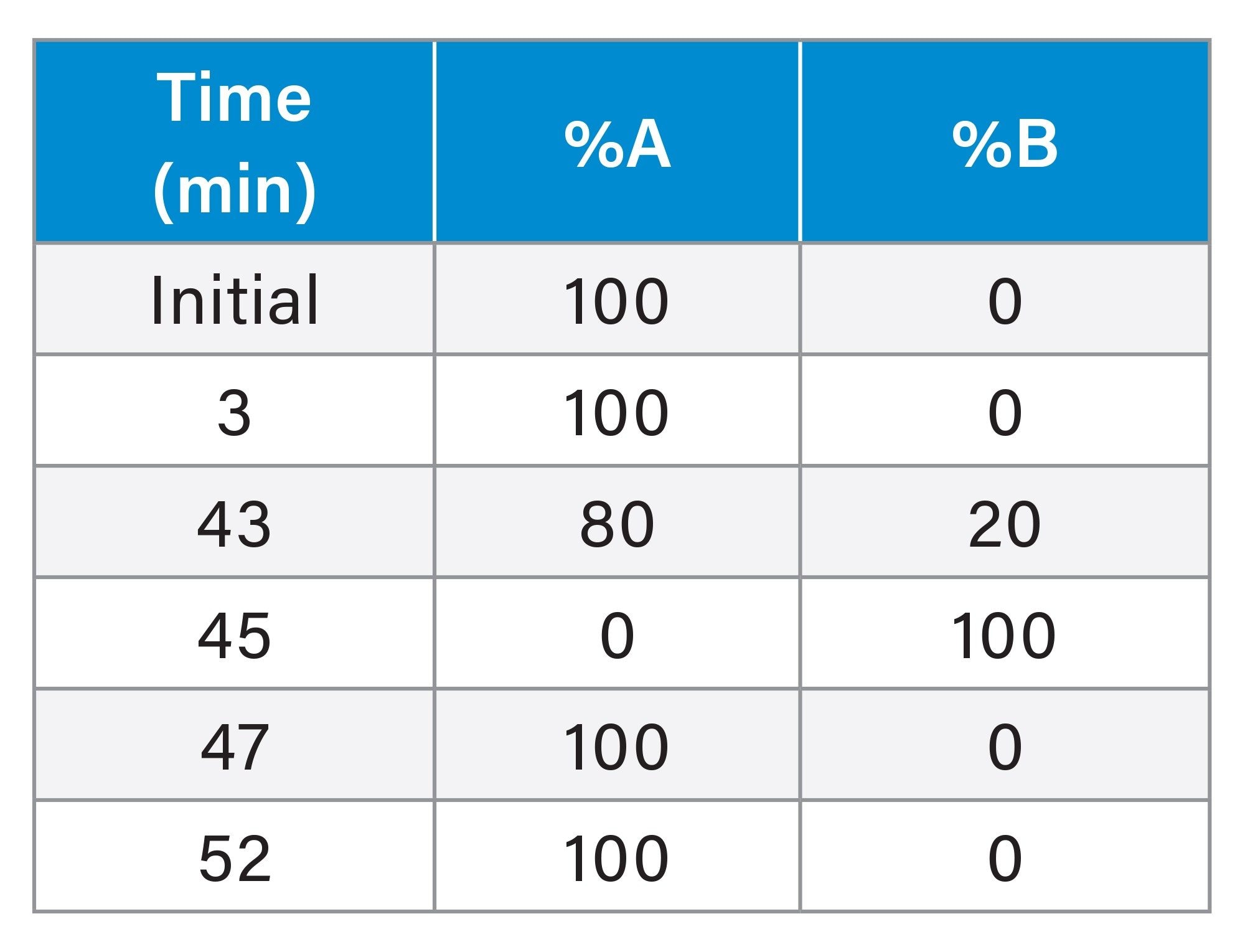
Purification Semi-Preparative IP-RP LC Method Conditions
|
HPLC system: |
Waters™ LC Prep AutoPurification System |
|
Column: |
XBridge Premier Oligonucleotide BEH C18 OBD Prep Column, 300Å, 2.5 µm, 10 X 100 mm (Custom packed) XBridge Premier Oligonucleotide BEH C18 300 Å, OBD Prep Column, 5µm, 10 X 100 mm (p/n: 186011170) |
|
Buffer: |
100 mM HAA, pH 7.0 (HA + acetic acid, pH adjusted) |
|
Mobile phase A: |
5% ACN in 100 mM HAA |
|
Mobile phase B: |
80% ACN in 100 mM HAA |
|
Gradient: |
Linear, 30–63,5% B over 47 minutes |
|
Flow rate: |
4.0 mL/min for 2.5 µm 8.0 mL/min for 5 µm |
|
Injection volume: |
1 mL (in buffer A) |
|
Mass load: |
1022.4 nmol |
|
Column temperature: |
60 ºC |
|
Detection: |
Waters 2489 UV/Vis Detector, 280 nm |
Table 4. Machine parameters for the development of the semi-preparative IP-RPLC method.
Results and Discussion
Small-Scale IP-RP Method Development
IP-RP LC of oligonucleotides relies on the use of ion-pairing additives in the mobile phase.3 These additives, typically alkylamines, adsorb onto the hydrophobic stationary phase and facilitate electrostatic interactions between the positively charged alkylamines and the negatively charged phosphate backbone of the oligonucleotides. While IP-RP LC provides effective resolution for shorter oligonucleotides in the 15–30 nucleotide (nt) range, the separation becomes increasingly challenging as oligonucleotide length increases. Resolving species in the 40–60 nt range is more difficult, and separation of oligonucleotides in the 60–100 nt range presents a significant challenge.3 This difficulty arises because the relative charge difference between closely sized oligonucleotides decreases with length, for example, a 10 nt vs. 11 nt oligonucleotide differs by approximately 10% in charge, whereas the difference between 100 and 101 nt is only about 1%, resulting in diminished separation selectivity.3
To enable effective separation of long RNA molecules, careful selection of both the chromatography column and ion-pairing reagent was essential. Several columns were evaluated during the initial screening, including 3 mL RPC 15 and C18 purification cartridges. These columns were assessed based on peak shape, resolution of closely eluting fragments, and robustness. However, from different suppliers very poor separation was observed. For this reason, an alternative was sought, namely an XBridge Premier Oligonucleotide BEH C18, OBD Prep Column 300 Å, 2.5 µm, 4.6 mm x 100 mm. This column was chosen for its high efficiency, pH stability, and wide sorbent pore size, making it well-suited for separation of 60-100 nt long RNA species.
When selecting an appropriate ion-pairing reagent, several criteria were considered: the reagent had to be readily available, exhibit low toxicity to laboratory personnel, and be cost-effective. While 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and N,N-dimethylbutylamine (DBA) are frequently used in IP-RP LC of oligonucleotides, their toxicity and cost posed significant drawbacks for routine use. As alternatives, triethylammonium acetate (TEAA) and hexylammonium acetate (HAA) were selected.
To streamline method development, previously published application notes4 were used as a foundation, eliminating the need to design the workflow from first principles. As part of this optimization, the acetonitrile (ACN) content in the B buffer was increased to enhance method versatility and provide greater flexibility during gradient development.
In addition to TEAA and HAA, ammonium acetate (NH₄OAc) was also evaluated as a potential mobile phase. However, it failed to provide retention for the target oligonucleotide and was therefore excluded from further consideration. Chromatographic results obtained using the TEAA modified mobile phase can be seen in Figure 3 and the parameters can be observed in Table 1 and Table 2. The HAA results can be seen in Figure 4 and the parameters in Table 3. The data indicates that HAA promotes a stronger interaction between the RNA and the stationary phase than TEAA, which can be attributed to the greater hydrophobicity of HAA5. Both ion-pairing agents delivered comparable overall performance; however, HAA achieved a higher resolution for the full-length product (FLP)-X fragments. As a result, the HAA method was selected for scaling up to preparative purification.
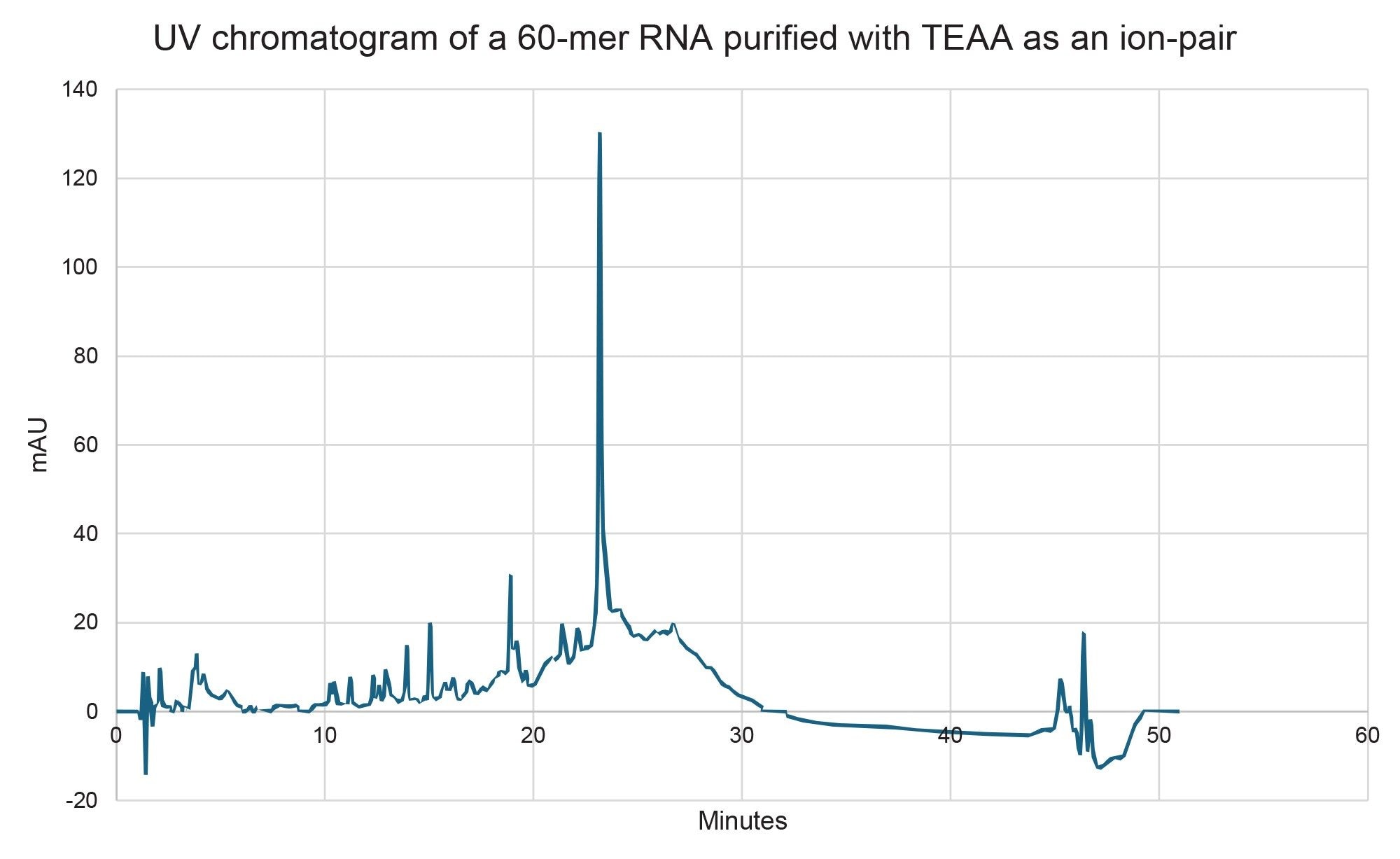
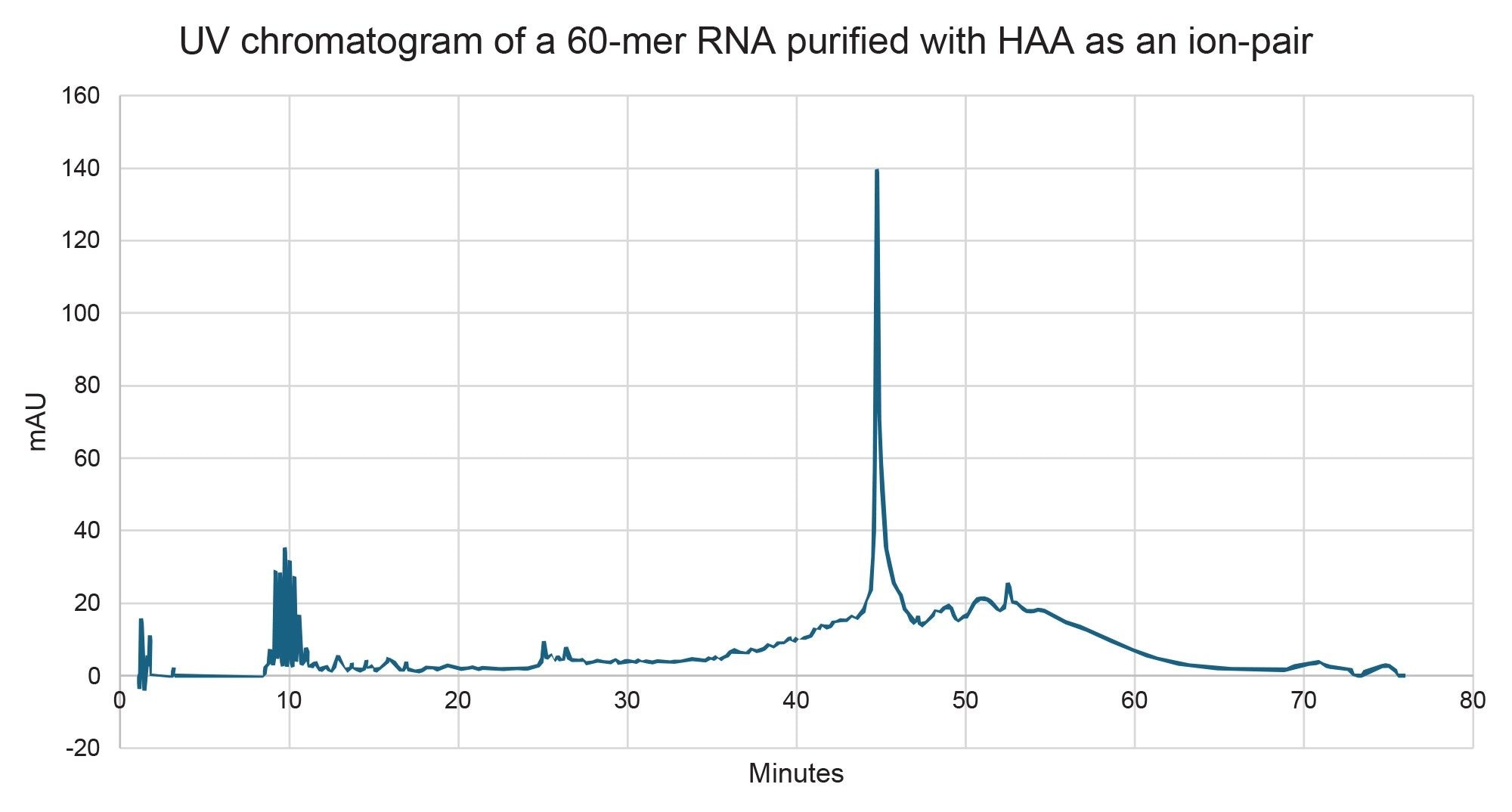
Prep HPLC IP-RP Method Development
Following the successful development of the small-scale method, the procedure was transferred to a preparative HPLC system for scale-up. To accommodate larger injection volumes, larger total mass loaded and increase throughput, the column diameter was scaled from 4.6 mm to 10 mm, while all other chromatographic parameters were kept the same.
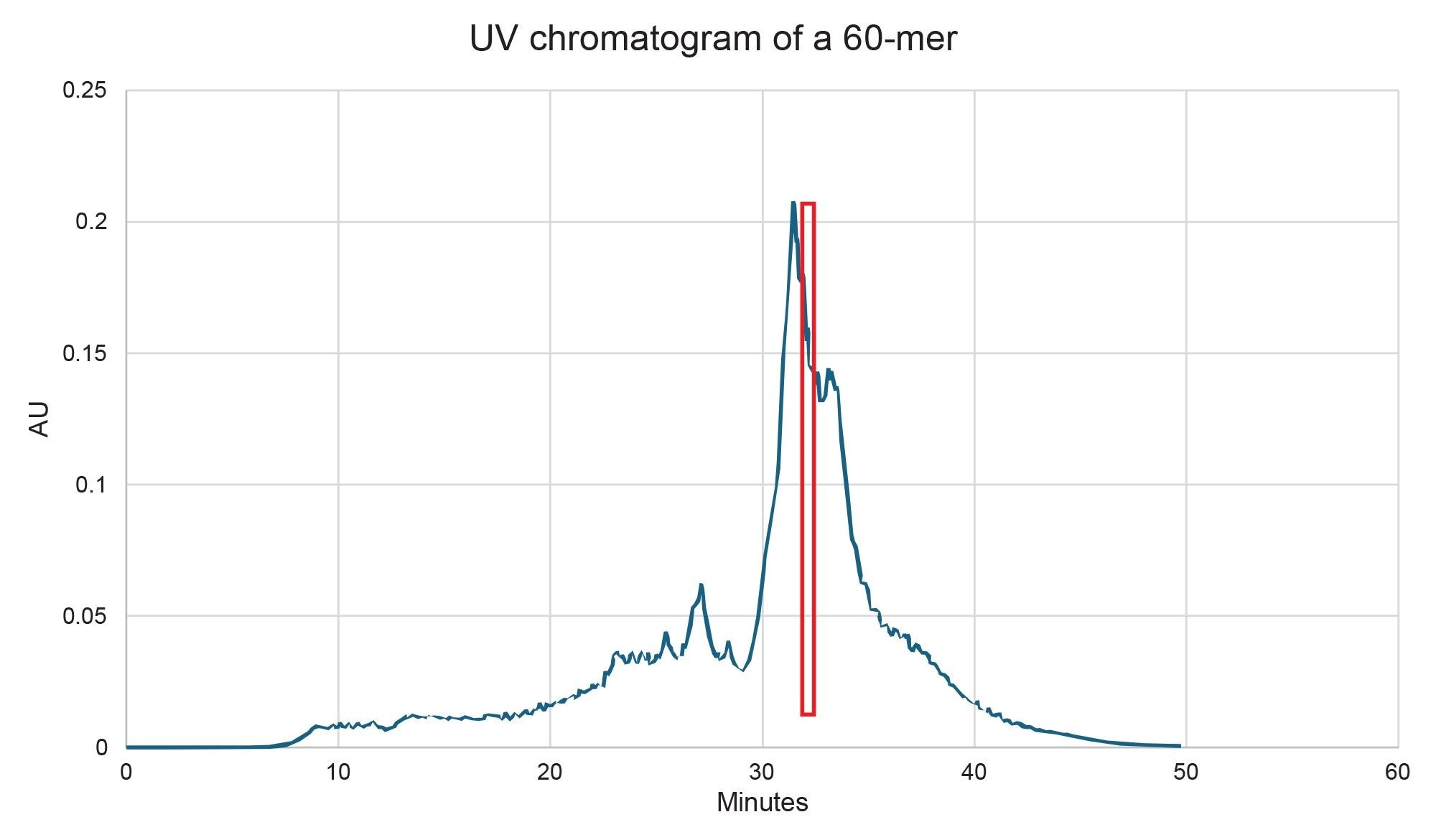
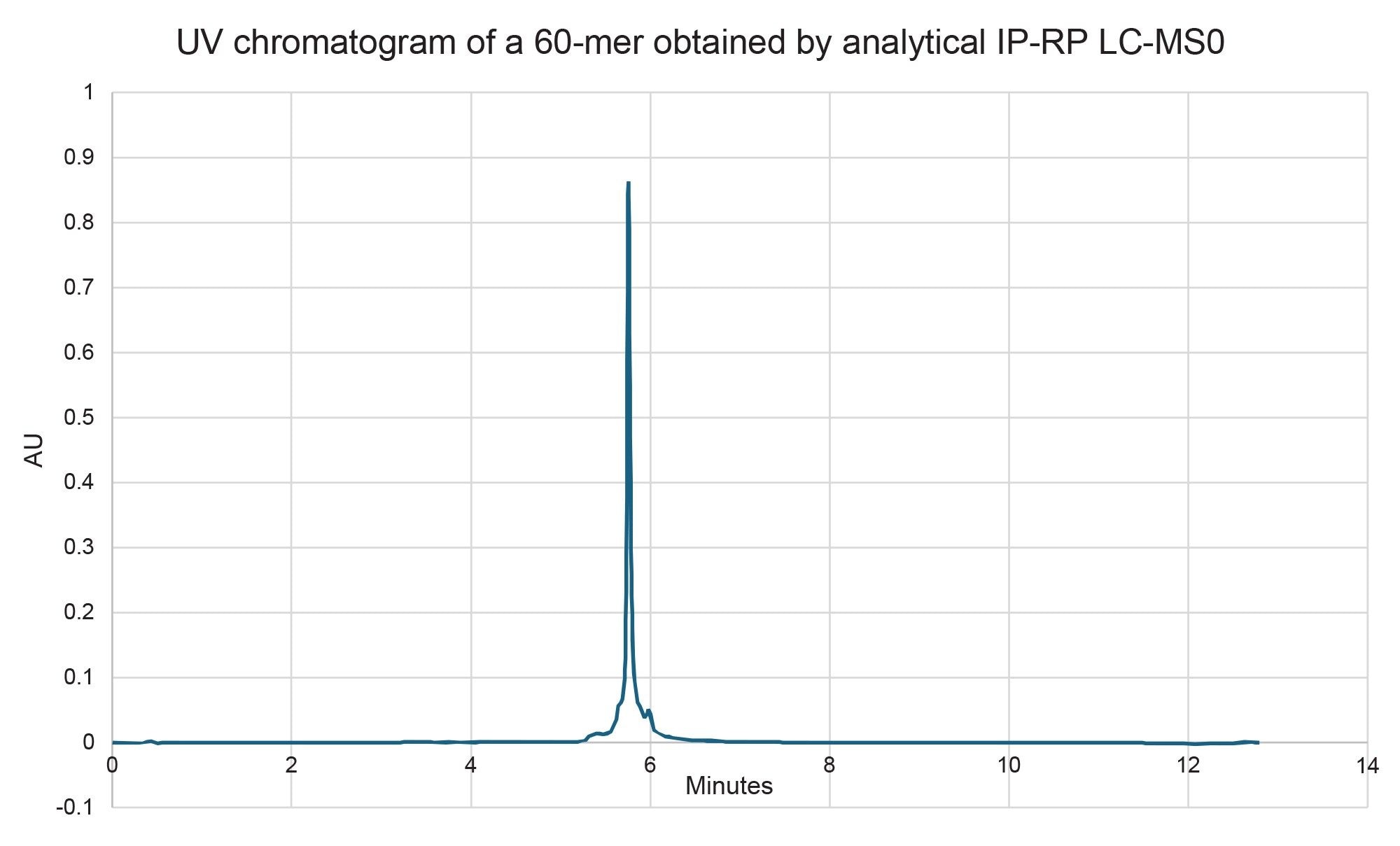
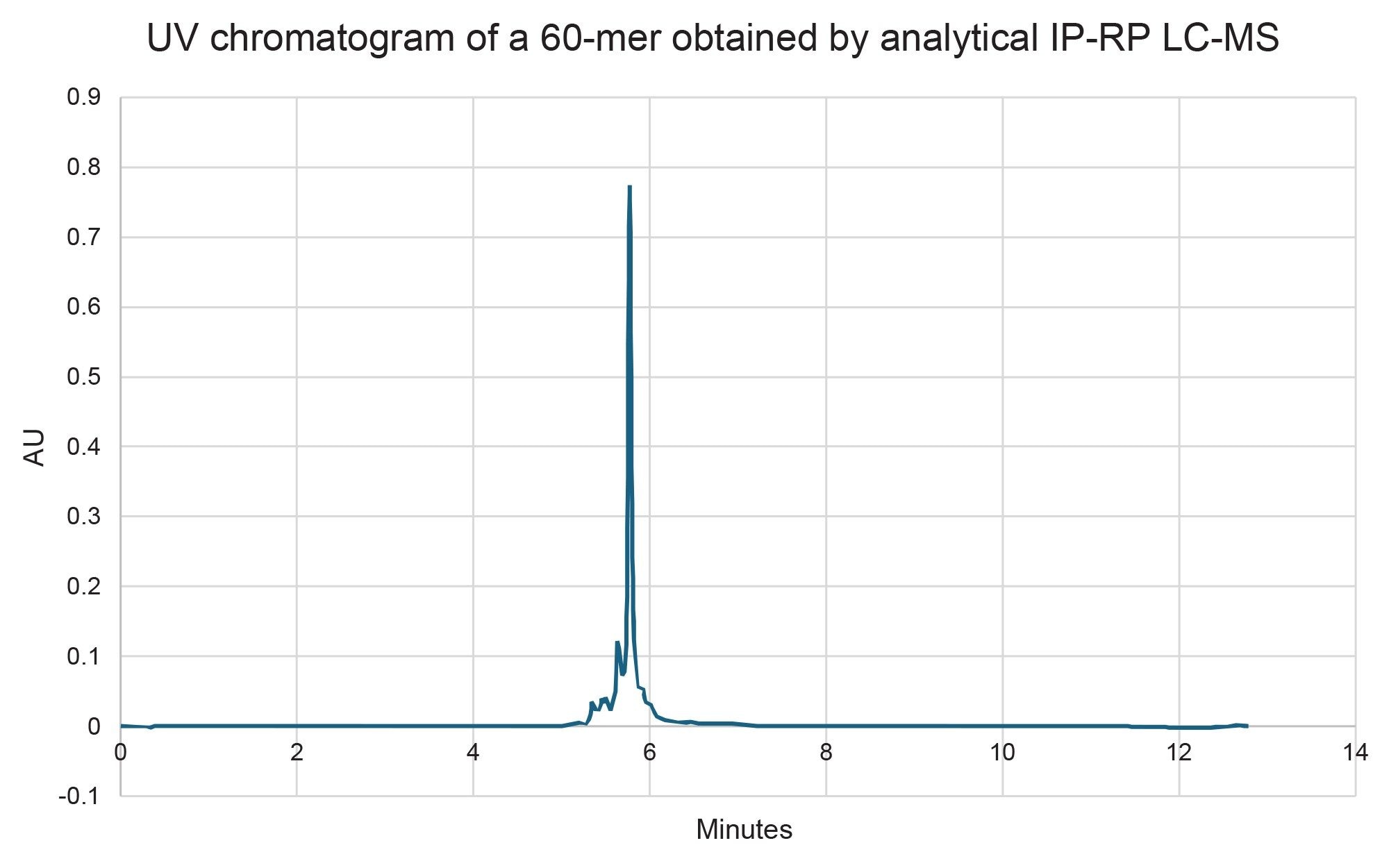
The scalability and system compatibility were evaluated for the 2.5 µm and 5 µm particle size versions of the 10 x 100 mm XBridge Premier Oligonucleotide BEH C18 300 Å, OBD Prep Columns. The 2.5 µm column, ran at 4 mL/min, generated a backpressure of approximately 3000 psi and achieved a final purity of 70% with a 4 mg yield of target RNA (Figure 5). The 5 µm column, on the other hand, produced the same backpressure (~3000 psi) at a higher flow rate of 8 mL/min, but delivered a lower purity of 62%, albeit in slightly higher 5 mg isolated yield (Figure 6). The purity values were determined by analytical ion-pair reversed-phase liquid chromatography mass spectrometry (IP-RP LC-MS) (Figure 7 for the 2.5 µm column and Figure 8 for the 5.0 µm column).
Importantly, while the 5 µm column did not offer an equal to or higher purity compared to the 2.5 µm column, it demonstrated compatibility with HPLC systems that have lower maximum pressure limits, making it a practical option in more pressure-sensitive setups. Attempts to decrease the pooled fractions on the 5 µm column to improve purity were unsuccessful, whereas the 2.5 µm column could be pushed to a 5 mg yield by pooling more fractions (albeit at a compromise to purity). These findings emphasize the trade-offs between resolution and hardware flexibility when selecting preparative column formats for long oligonucleotide purification.
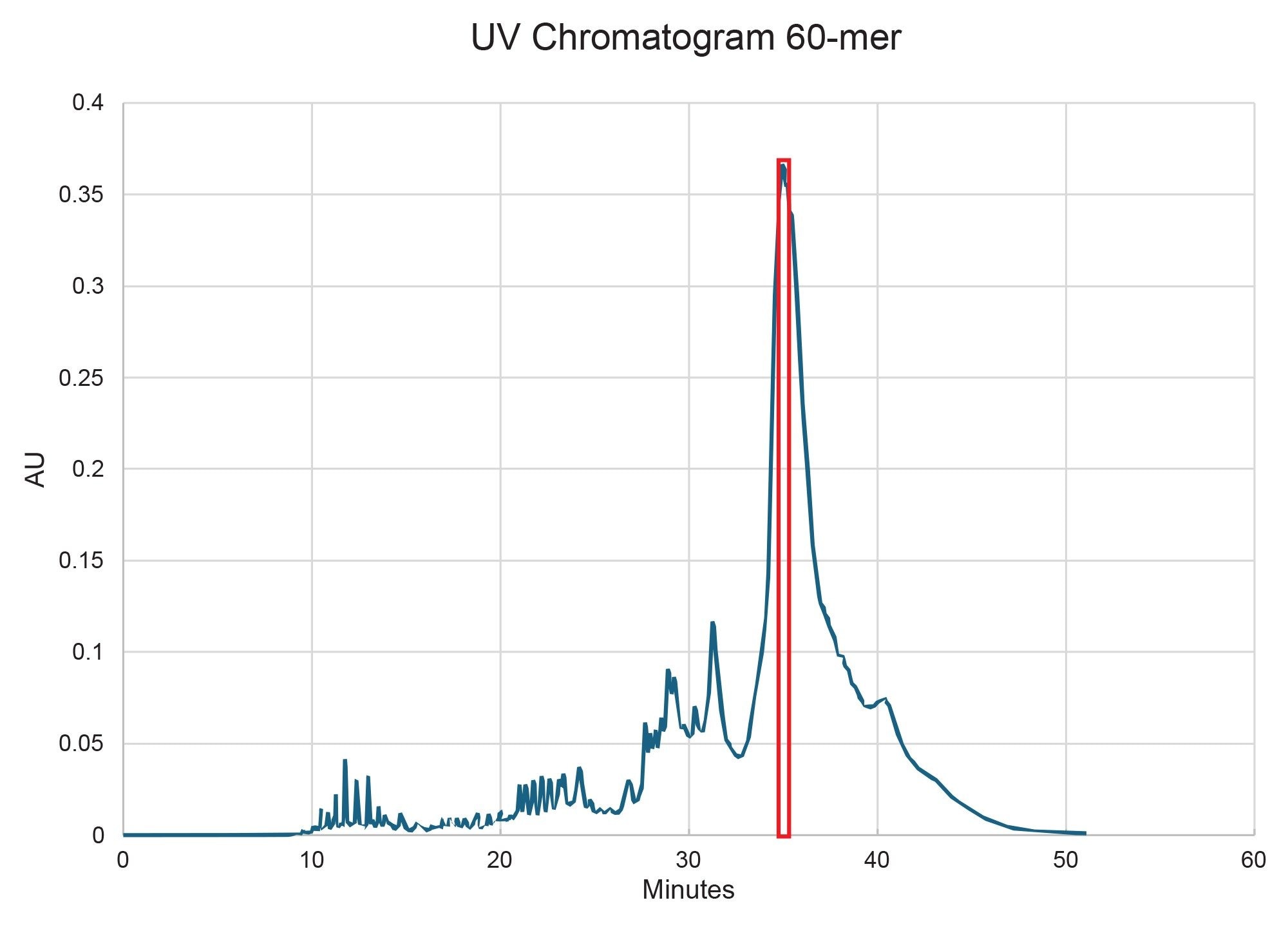
Conclusion
This study demonstrates how advanced technologies such as Waters MaxPeak High Performance Surfaces (HPS) can be effectively applied in preparative oligonucleotide chromatography to overcome longstanding challenges in the purification of long synthetic RNA sequences. Traditional stainless-steel hardware can introduce undesirable interactions, especially during early purification cycles, potentially compromising purity, recovery, and the reliability of downstream toxicology assessments. In contrast, MaxPeak HPS Columns offer inert, organosilica-protected surfaces that minimize these interactions and ensure consistent retention and recovery starting from the first injection.
A systematic approach to method development was employed, beginning with the screening of multiple columns and ion-pairing reagents where TEAA, HAA, and ammonium acetate were tested. Only TEAA and HAA delivered suitable performance, with HAA providing enhanced retention and superior resolution for FLP-X fragments. Consequently, the HAA-based method was selected for scale-up.
On the column selection front, several columns were evaluated. The MaxPeak Premier Oligonucleotide BEH C18 Columns with 300 Å pore size consistently outperformed other tested columns in terms of resolution and robustness, making them the basis of the final method.
Preparative purifications were carried out using both 2.5 µm and 5 µm particle size columns (10 × 100 mm). While the 2.5 µm column achieved a final purity of 70% with a 4 mg RNA yield at 3000 psi and 4 mL/min flow rate, the 5 µm column operating at the same pressure but at 8 mL/min yielded 5 mg of RNA with reduced purity (62%).
In summary, this work underscores the importance of intelligent method design including informed reagent and hardware selection with performance-enhancing technologies such as MaxPeak HPS to develop scalable, high-resolution purification methods for long RNA oligonucleotides.
Waters, MaxPeak, XBridge, BEH and OBD are trademarks of Waters Technologies Corporation. Mermade is a trademark of LGC Genomics Limited. UltiMate is a trademark of Dionex Corporation.
References
- Mitigation of analyte loss on metal surfaces in liquid chromatography. Gilar, M., DeLano, M., & Gritti, F. 462247, s.l. : Journal of Chromatography A, 2021, Vol. 1650.
- Technical Brief - Procedure for the Synthesis and Deprotection of Synthetic RNA. research, Glen. s.l. : Glen research, Vol. Glen Report 19.22.
- Ion-Pair Reversed-Phase Liquid Chromatography Method for Analysis of mRNA Poly(A) Tail Heterogeneity. Martin Gilar, Maissa M. Gaye. 2023, Waters Application note, 720007873.
- HPLC Purification OF Long Synthetic Oligonucleotides. Waters. WA31789. 2003.
- Effect of ion-pairing reagent hydrophobicity on liquid chromatography and mass spectrometry analysis of oligonucleotides. M. Donegan, J.M. Nguyen, and M. Gilar. s.l. : elsevier, 2022, Vol. 1666.
720009065, October 2025