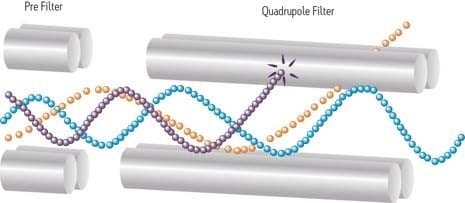
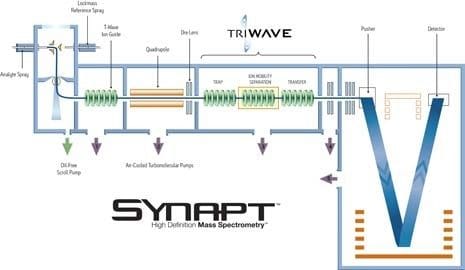
An ion trap instrument operates on principles similar to those of a quadrupole instrument. Unlike the quadrupole instrument, however, which filters streaming ions, both the ion trap and more capable ion cyclotron (ICR) instrument store ions in a three-dimensional space. Before saturation occurs, the trap or cyclotron allows selected ions to be ejected, according to their masses, for detection. A series of experiments can be performed within the confines of the trap, fragmenting an ion of interest to better define the precursor by its fragments. Fields generated by RF voltages applied to a stacked or "sandwich" geometry (end-cap electrodes at opposing ends) trap ions in space between the two electrodes. Ramping or scanning the RF voltage ejects ions from their secular frequency, or trapped condition. Dynamic range is sometimes limited. The finite volume and capacity for ions limits the instrument's range, especially for samples in complex matrices.
Ion trap instruments were introduced in the 1980s. But limitations imposed by the internal ionization scheme used in those early instruments prevented their use for many applications. Only with the advent of external ionization did the instruments become more universally practical.
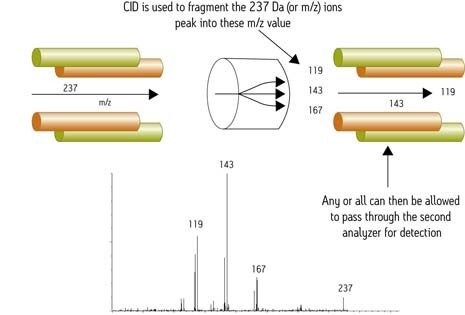
The ability to perform sequential fragmentation and thus derive more structural information from a single analyte (i.e., fragmenting an ion, selecting a particular fragment, and repeating the process) is called MSn. GC chromatographic peaks are not wide enough to allow more than a single fragmentation (MS/MS or MS2). Ion trap instruments perform MS/MS or fragmentation experiments in time rather than in space, like quadrupole and sector instruments. So they cannot be used in certain MS/MS experiments like neutral loss and precursor ion comparisons. Also, in MS/MS operation with an ion trap instrument, the bottom third of the MS/MS spectrum is lost, a consequence of trap design. To counter the loss, some manufacturers make available via their software wider scan requirements that necessitate the switching of operating parameters during data acquisition,
The trap design places an upper limit on the ratio between a precursor's mass-to-charge ratio (m/z) and the lowest trapped fragment ion, commonly known as the "one third rule". For example, fragment ions from an ion at m/z 1500 will not be detected below m/z 500 - a significant limitation for the de novo sequencing of peptides. The ion trap has limited dynamic range, the result of space-charge effects when too many ions enter the trapping space. Manufacturers have developed automated scanning, which counts ions before they enter the trap, limiting, or gating, the number allowed in. Difficulty can still be encountered when a relatively small amount of an ion of interest is present in a large population of background ions.
Because of similarities in functional design, quadrupole instruments are hybridized to incorporate the advantages of streaming quadrupole and ion trapping behavior to improve sensitivity and allow on-the-fly experiments not possible with either alone. Such instruments are sometimes called linear traps or Q-traps). The increased volume of a linear trap instrument (over a three-dimensional ion trap) improves dynamic range.
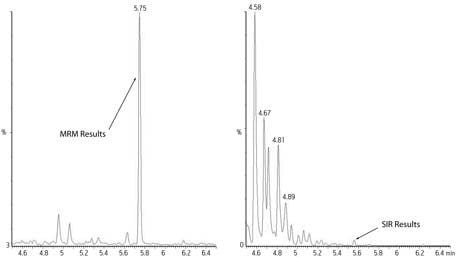
Ion trap instruments do not scan as a quadrupole instrument does so using the single ion monitoring (SIM), or single ion recording (SIR), technique does not improve sensitivity on ion traps as it does on quadrupole and sector instruments.
Fast-fourier transform ion cyclotrons (FTICR) represent the extreme capability of measuring mass with the ability to resolve closely related masses. Although impractical for most applications, a 14.5-tesla magnet can achieve a resolution of more than 3.5 million and thus display the difference between molecular entities whose masses vary by less than the mass of a single electron.
Cyclotron instruments trap ions electrostatically in a cell using a constant magnetic field. Pulses of RF voltage create orbital ionic motion, and the orbiting ions generate a small signal at the detection plates of the cell (the ion's orbital frequency). The frequency is inversely related to the ions' m/z, and the signal intensity is proportional to the number of ions of the same m/z in the cell. At very low cell pressures, a cyclotron instrument can maintain an ion's orbit can for extended periods providing very high resolution measurements.
Sustained off-resonance, irradiation, collision-induced dissociation (SORI-CID) is a CID technique used in Fourier-transform ion cyclotron resonance mass spectrometry. The ions are accelerated in cyclotron motion where increasing pressure results in collisions that produce fragments. After the fragmentation, the pressure is reduced and the high vacuum restored to analyze the fragment ions.
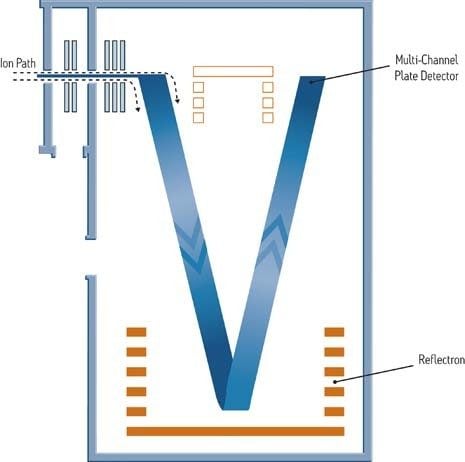
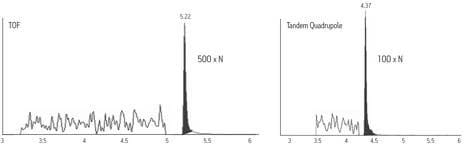
Time-of-flight (TOF) instruments, although developed many years ago, have become the basis for much modern work because of their fast, precise electronics and modern ionization techniques like ESI. A TOF instrument provides accurate mass measurement to within a few parts-per-million (ppm) of a molecule's true mass. A temporally dispersive mass analyzer, the TOF instrument is used in a linear fashion or, aided by electrostatic grids and lenses, as a reflectron. When operated as a reflectron, resolution is increased without dramatically losing sensitivity or needing to increase the size of the flight (or drift) tube.