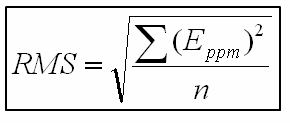Comparing precision from instrument to instrument: millimass units (mmu), measurement error (ppm), and resolution
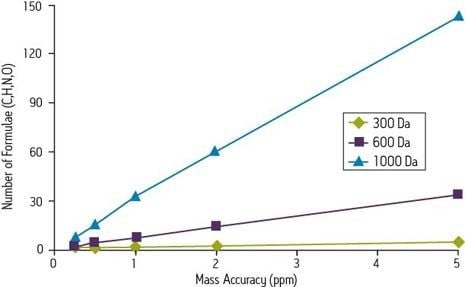
According to the Accurate Mass Best Practice Guide of the VIMMS Programme, an initiative that forms part of the UK National Measurement System, most instruments used for accurate mass measurements are capable of achieving precision of 10 ppm or better.
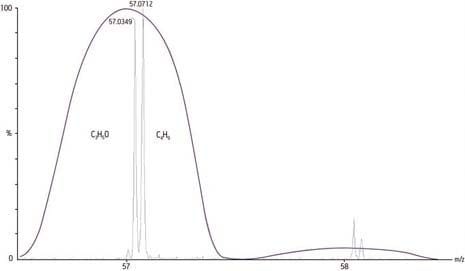
A calculated mass of 118 Da measured by a modern mass spectrometer to within 2 mmu accuracy would display 17 ppm error, sufficient by today’s standards for unambiguous determination of a chemical formula of that mass:
Monoisotopic calculated exact mass = 118 Da
Measured accurate mass = 118.002 Da
Difference = 0.002 mmu
Error [Difference/exact mass x 106] = 17 ppm
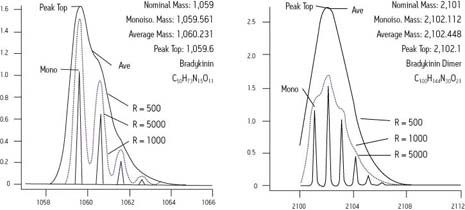
An instrument capable of a response at 750 m/z, also deficient by 2 mmu, would be accurate to 2.7 ppm. In the first case, the measurement is more than sufficient for unambiguous identification of a chemical formula, according to the published standards of the Journal of The American Society for Mass Spectrometry. But in the latter case, the measurement is insufficiently precise. Only the highest order Fourier transform ion cyclotron resonance mass spectrometry (FTICR) can achieve such precision at higher masses.
A comprehensive method of evaluating instrument mass accuracy measurement capability which resembles intended use is by calculating the root mean square or RMS error. To illustrate its use, the following is adapted from the mass measurement accuracy specification of a commercial TOF mass spectrometer.
"The mass measurement accuracy of the instrument, under normal operating conditions, will be better than a given ppm RMS over the given m/z range, based on a number of consecutive repeat measurements of an analyte peak (of given m/z), using a suitable reference peak (of given m/z). Analyte and reference peaks must have sufficient intensity and be free of interference from other masses."
There are some important points and assumptions need to be considered:
- An instrument calibration has already been performed with peaks of known mass using a calibration standard. The reference peak is used to account for any variation in the instrument calibration over time and mass measurement accuracy is determined using the analyte peak.
- Normal operating conditions - can also include details of chromatographic conditions (for LC-MS performance specifications) and any related MS operating conditions (e.g. mass resolution, m/z of interest, or spectral acquisition rate).
- Sufficient intensity - assumes that the ion count is not detrimental to characterization of the (mass measurement) accuracy and precision of the instrument in question. Too few ions leads to poor ion statistics and too many ions can lead to detector saturation, both of which result in a greater variation in the standard deviation of repeat measurements and will adversely influence calculation of the RMS error (also relevant to instrument calibration).
- Free from interferences - assumes that the mass measurement of the peak of known mass is free from interference by ions of the same or similar mass. Overlapping peaks lead to poor mass measurement accuracy which is also detrimental to properly characterizing the accuracy or precision of the instrument (also relevant to instrument calibration).
- The reference is a good representation of the m/z range which is relevant to the analysis of a particular sample type.
The RMS error is calculated using the following relation, where Eppm is the ppm error, and n is the number of masses considered: