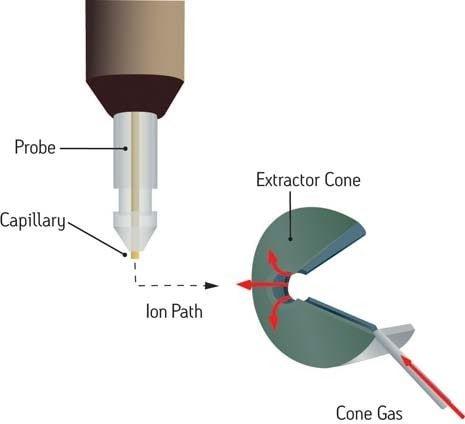
Chemical Ionization (CI)
Molecules that fragment excessively call for "soft" techniques. Chemical ionization (CI) produces ions by a gentler proton transfer process that preserves and promotes the appearance of the molecular ion itself. The sample is exposed to an excess of reagent gas such as that which evolves when methane forms the protonated molecular ion (M+H). The reverse process can produce negative ions. Transferring the proton to the gas molecule can, in some cases, produce the negative ion (M-H).
Chemical Ionization (CI) is sometimes used for compounds with chemistry similar to those analyzed by EI to enhance the abundance or appearance of the molecular ion in favor of significant fragmentation. Similar to EI, samples must be thermally stable since heating in the source causes vaporization. The ionization mechanism of CI relies on EI for the initial ionization step but within the source is a chemical reagent gas, such as methane, isobutane or ammonia, at high pressure. The reagent gas, which is present at a much higher concentration than the analyte (R), is ionized by electron ionization to give primary R+. reagent ions. The collision of the R+. ions with neutral R molecules lead to the formation of stable secondary ions which are the reactant species which then ionize analyte molecules (A) by ion-molecule reactions.
For example the ion-molecule reaction between a methane ion and a methane molecule gives rise to the fairly stable CH5+ species.
CH4+. + CH4 --> CH5+ + CH3.
The reactant ion CH5+ can ionize neutral analyte molecules (A) by proton transfer, hydride abstraction or charge exchange.
RH+ + A --> R + AH+ (proton transfer)
(R-H)+ + A --> R + (A-H)+ (hydride abstraction)
R+. + A --> R + A+. (charge exchange)
The most common ionization reactions are protonation, which is favored for molecules with proton affinities higher than the reagent. Hydride abstraction is common for lower proton affinity molecules and charge exchange occurs with reagents of high ionization energy.
The substance to be analyzed is at a much lower pressure than the reagent gas. If we consider methane as the reagent gas the electron impact causes mainly ionization of the methane. This fragments in part to CH3+. These species then undergo ion molecule reactions under the high source pressures employed.
CH4+. + CH4 --> CH5+ + CH3.
CH3+ + CH4 --> C2H5+ + H2
CH5+ can act as a Bronsted acid and C2H5+ as a Lewis acid to produce ions from the analyte.
Careful choice of the CI reagent gas can improve charge transfer to an analyte molecule as the gas phase acidity of the chemical ionization gas influences the efficiency of the charge transfer. In CI the analyte is more likely to result in a molecular ion with the reduced fragmentation conserving the energy normally internalized in EI to break bonds.